

Clinical impact of the subclonal architecture and mutational complexity in chronic lymphocytic leukemia
- PMID: 28924241
- PMCID: PMC5843898
- DOI: 10.1038/leu.2017.291
Clinical impact of the subclonal architecture and mutational complexity in chronic lymphocytic leukemia
Abstract
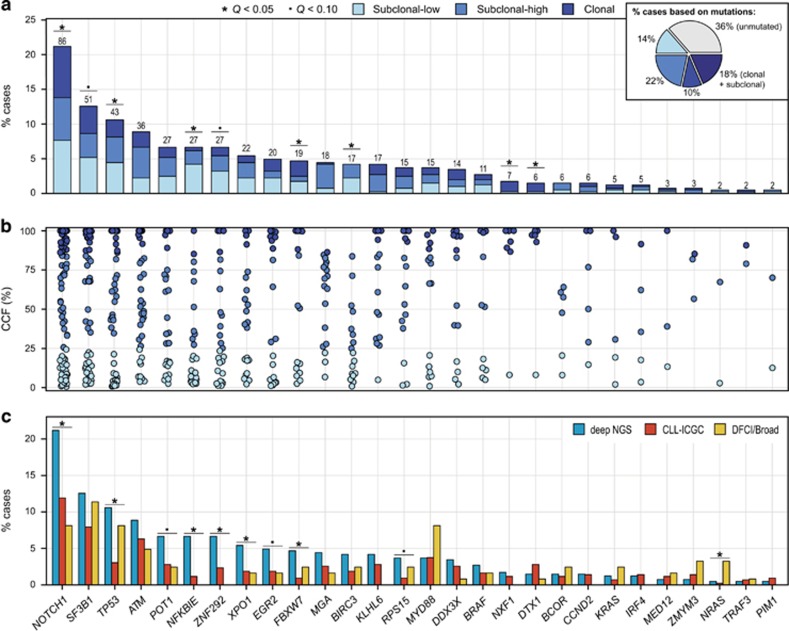
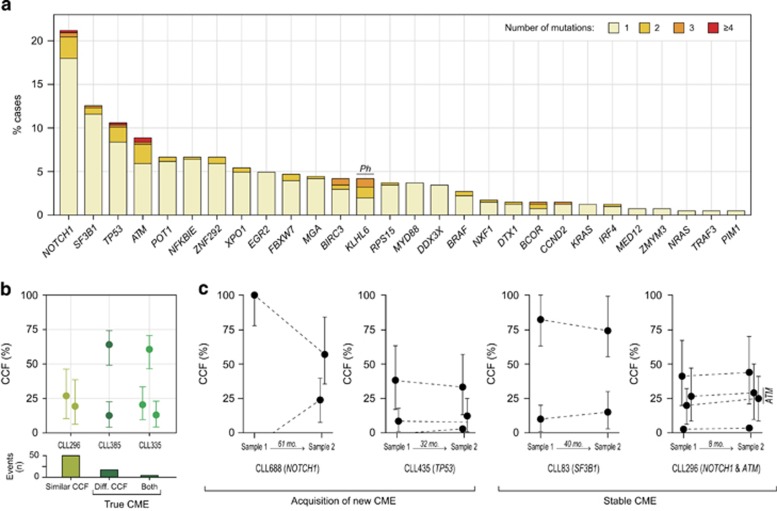
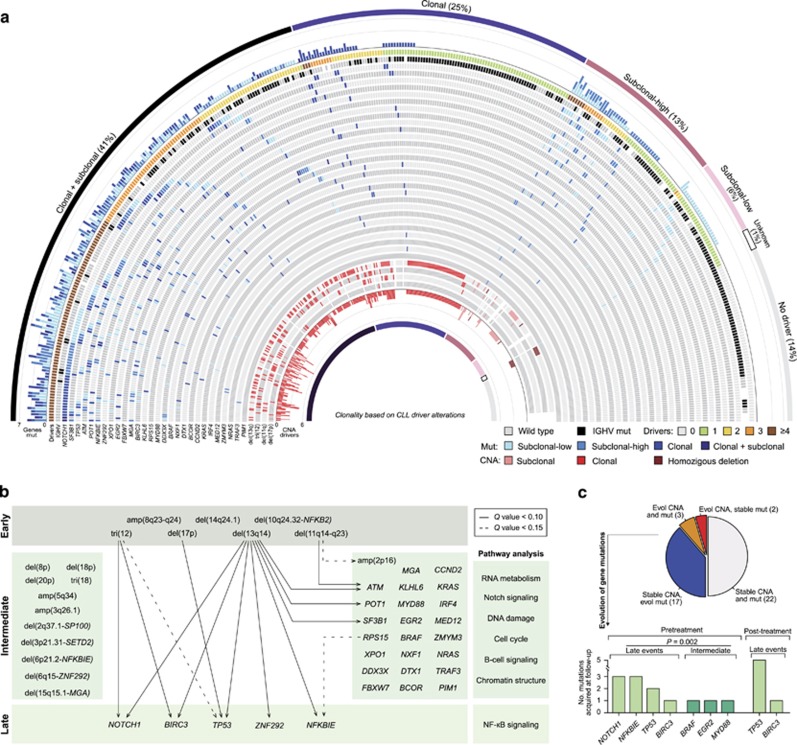
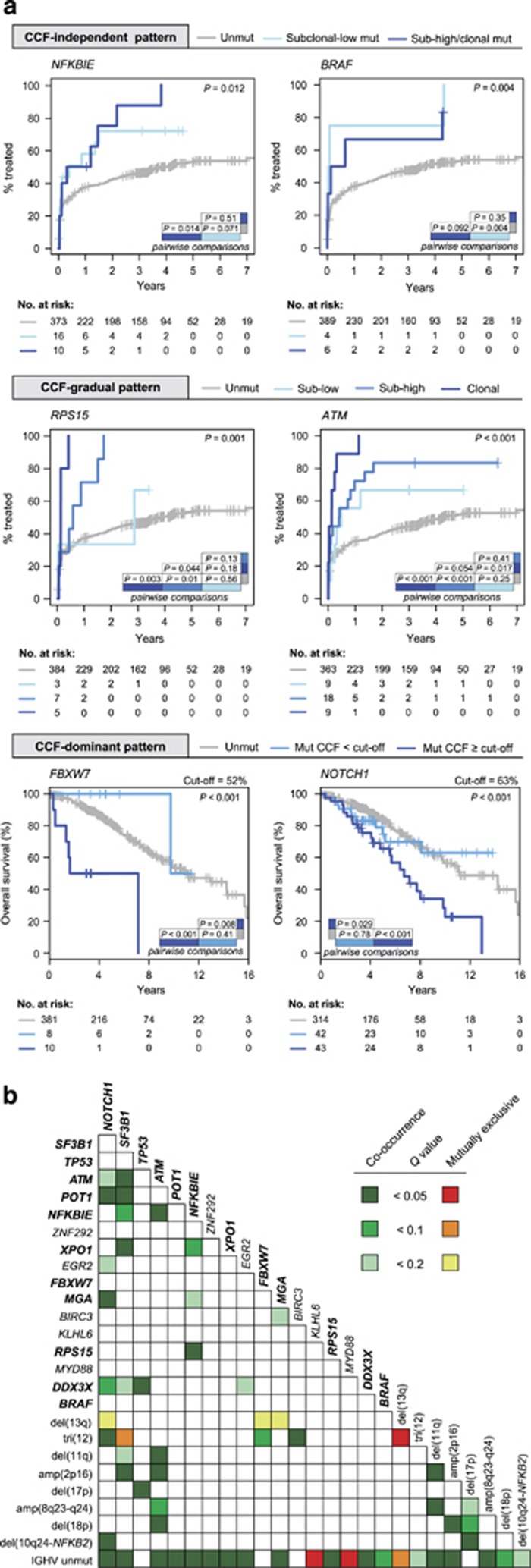
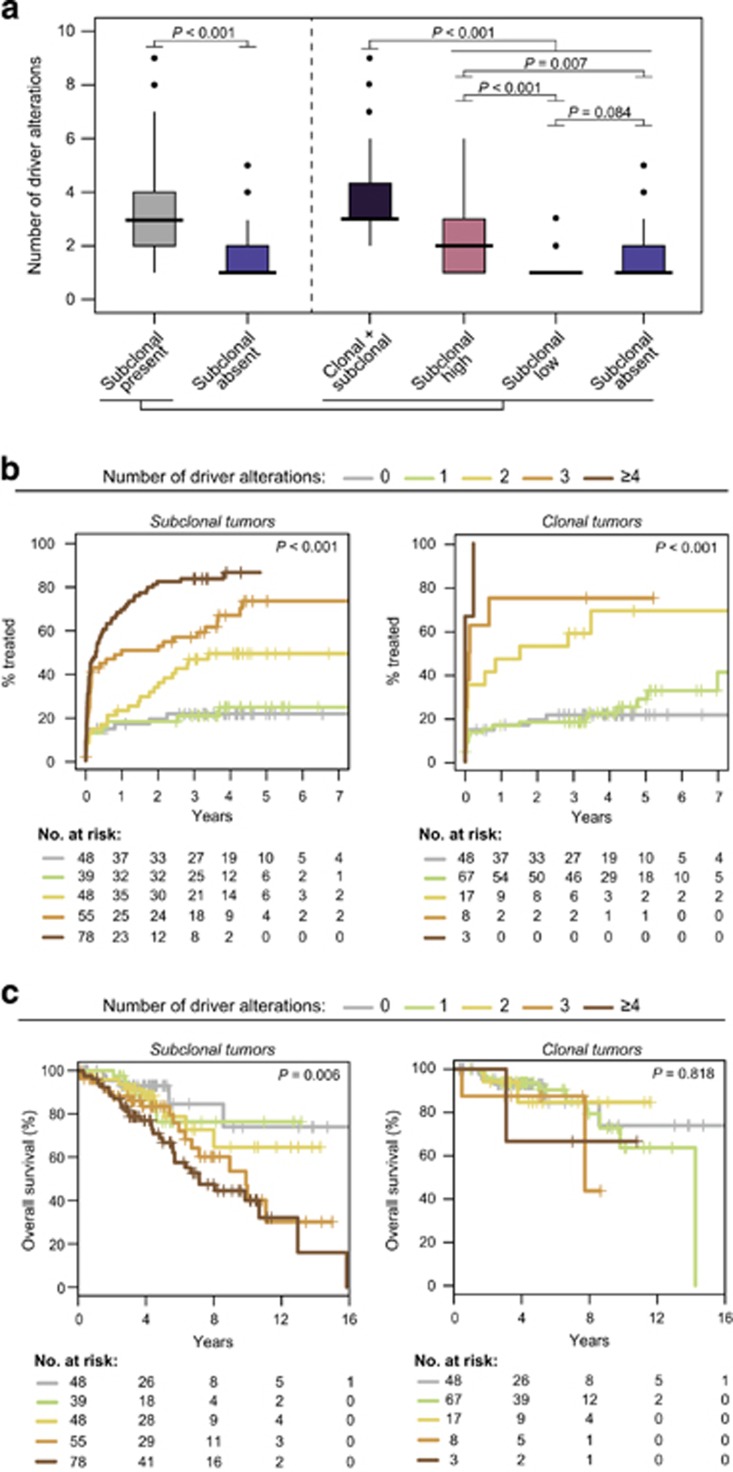
Genome studies of chronic lymphocytic leukemia (CLL) have revealed the remarkable subclonal heterogeneity of the tumors, but the clinical implications of this phenomenon are not well known. We assessed the mutational status of 28 CLL driver genes by deep-targeted next-generation sequencing and copy number alterations (CNA) in 406 previously untreated patients and 48 sequential samples. We detected small subclonal mutations (0.6-25% of cells) in nearly all genes (26/28), and they were the sole alteration in 22% of the mutated cases. CNA tended to be acquired early in the evolution of the disease and remained stable, whereas the mutational heterogeneity increased in a subset of tumors. The prognostic impact of different genes was related to the size of the mutated clone. Combining mutations and CNA, we observed that the accumulation of driver alterations (mutational complexity) gradually shortened the time to first treatment independently of the clonal architecture, IGHV status and Binet stage. Conversely, the overall survival was associated with the increasing subclonal diversity of the tumors but it was related to the age of patients, IGHV and TP53 status of the tumors. In conclusion, our study reveals that both the mutational complexity and subclonal diversity influence the evolution of CLL.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Quesada V, Conde L, Villamor N, Ordóñez GR, Jares P, Bassaganyas L et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet 2011; 44: 47–52. - PubMed
-
- Puente XS, Beà S, Valdés-Mas R, Villamor N, Gutiérrez-Abril J, Martín-Subero JI et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature 2015; 526: 519–524. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
