

Topological organization and dynamic regulation of human tRNA genes during macrophage differentiation
- PMID: 28931413
- PMCID: PMC5607496
- DOI: 10.1186/s13059-017-1310-3
Topological organization and dynamic regulation of human tRNA genes during macrophage differentiation
Abstract
Background: The human genome is hierarchically organized into local and long-range structures that help shape cell-type-specific transcription patterns. Transfer RNA (tRNA) genes (tDNAs), which are transcribed by RNA polymerase III (RNAPIII) and encode RNA molecules responsible for translation, are dispersed throughout the genome and, in many cases, linearly organized into genomic clusters with other tDNAs. Whether the location and three-dimensional organization of tDNAs contribute to the activity of these genes has remained difficult to address, due in part to unique challenges related to tRNA sequencing. We therefore devised integrated tDNA expression profiling, a method that combines RNAPIII mapping with biotin-capture of nascent tRNAs. We apply this method to the study of dynamic tRNA gene regulation during macrophage development and further integrate these data with high-resolution maps of 3D chromatin structure.
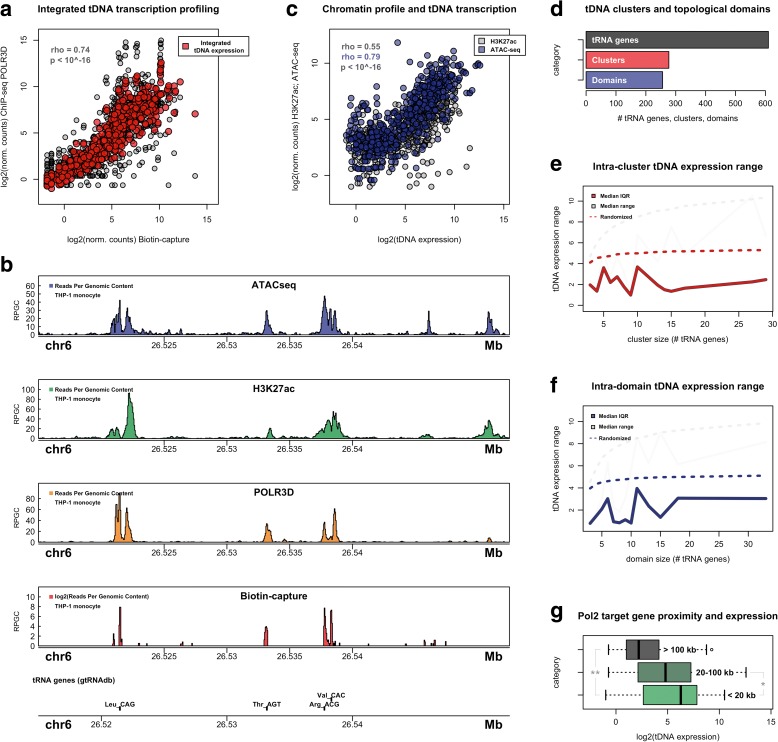
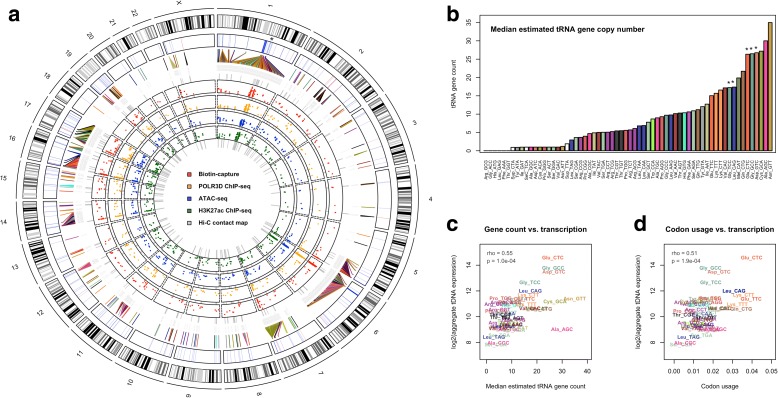
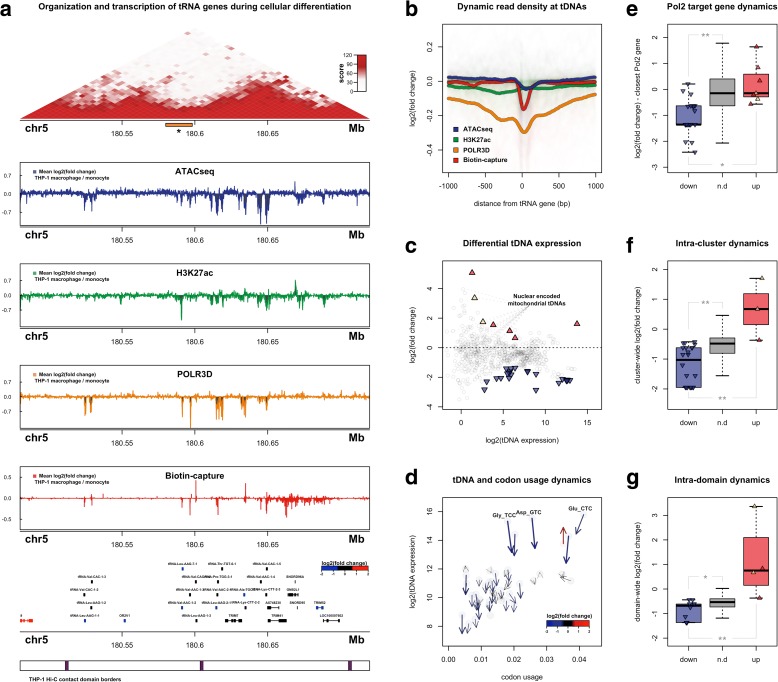
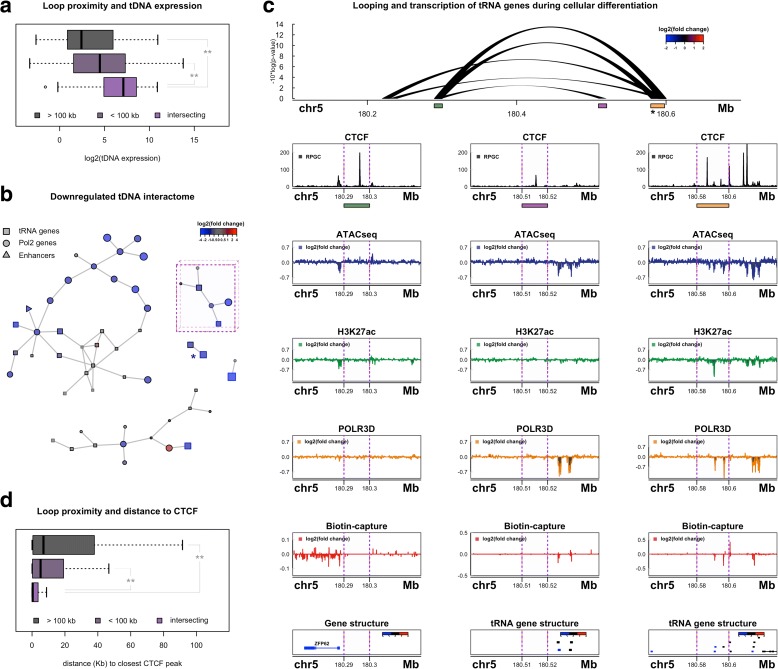
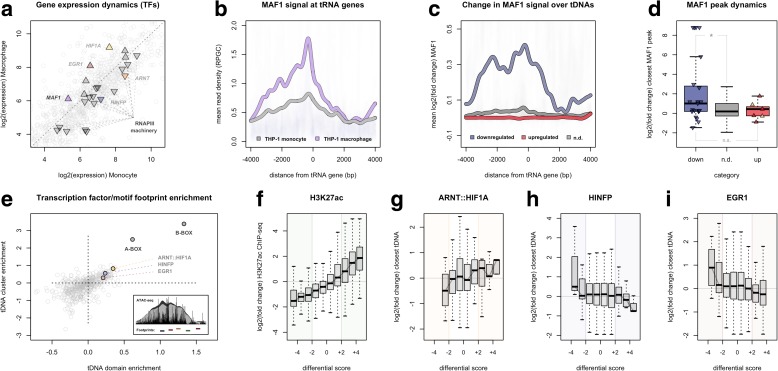
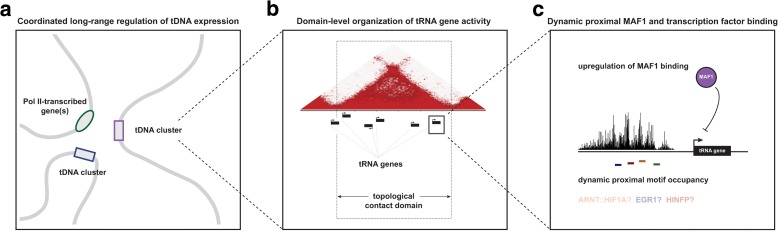
Results: Integrated tDNA expression profiling reveals domain-level and loop-based organization of tRNA gene transcription during cellular differentiation. tRNA genes connected by DNA loops, which are proximal to CTCF binding sites and expressed at elevated levels compared to non-loop tDNAs, change coordinately with tDNAs and protein-coding genes at distal ends of interactions mapped by in situ Hi-C. We find that downregulated tRNA genes are specifically marked by enhanced promoter-proximal binding of MAF1, a transcriptional repressor of RNAPIII activity, altogether revealing multiple levels of tDNA regulation during cellular differentiation.
Conclusions: We present evidence of both local and coordinated long-range regulation of human tDNA expression, suggesting the location and organization of tRNA genes contribute to dynamic tDNA activity during macrophage development.
Keywords: Biotin-capture; CTCF; Hi-C; TFIIIC; Topologically associating domains; tDNA; tRNAome.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Competing interests
MPS is a founder and member of the science advisory board of Personalis and Qbio and a science advisory board member of Genapsys and Epinomics.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
