

ALG9-CDG: New clinical case and review of the literature
- PMID: 28932688
- PMCID: PMC5596360
- DOI: 10.1016/j.ymgmr.2017.08.004
ALG9-CDG: New clinical case and review of the literature
Abstract
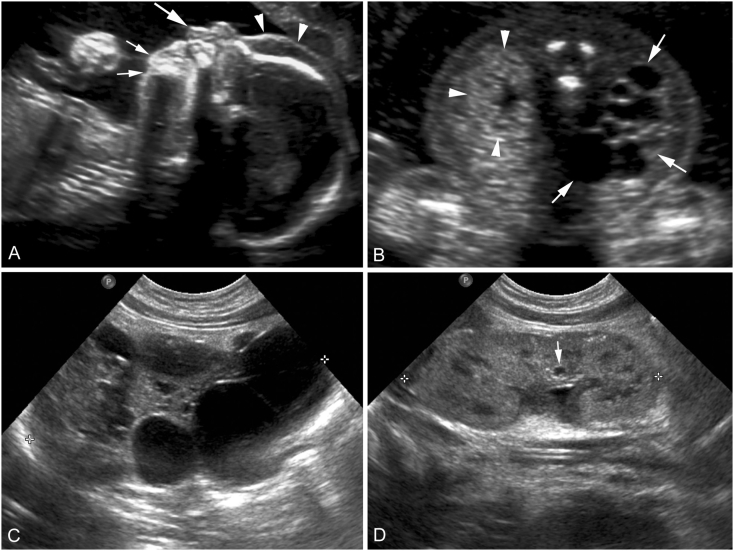
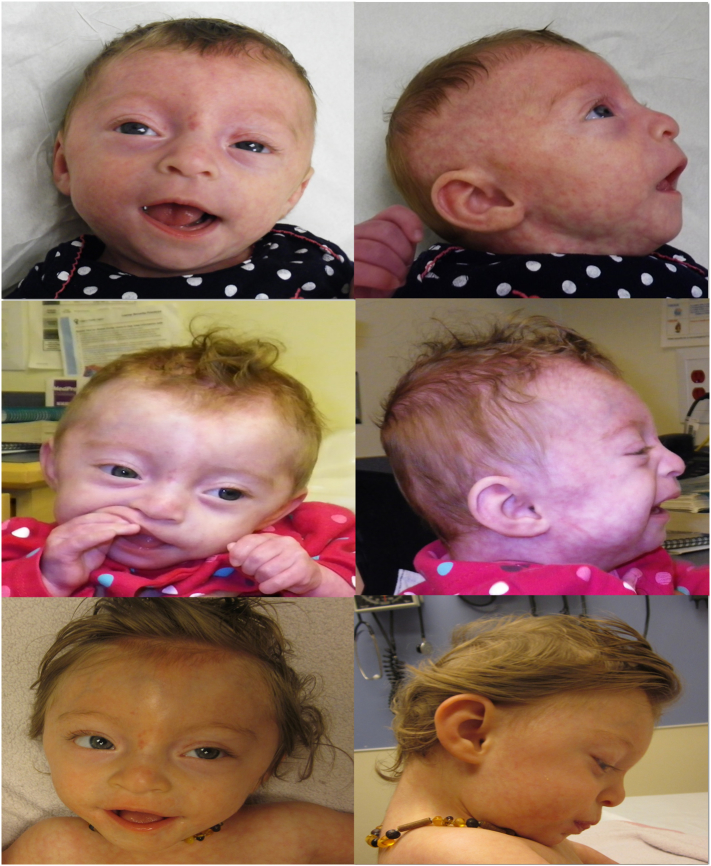
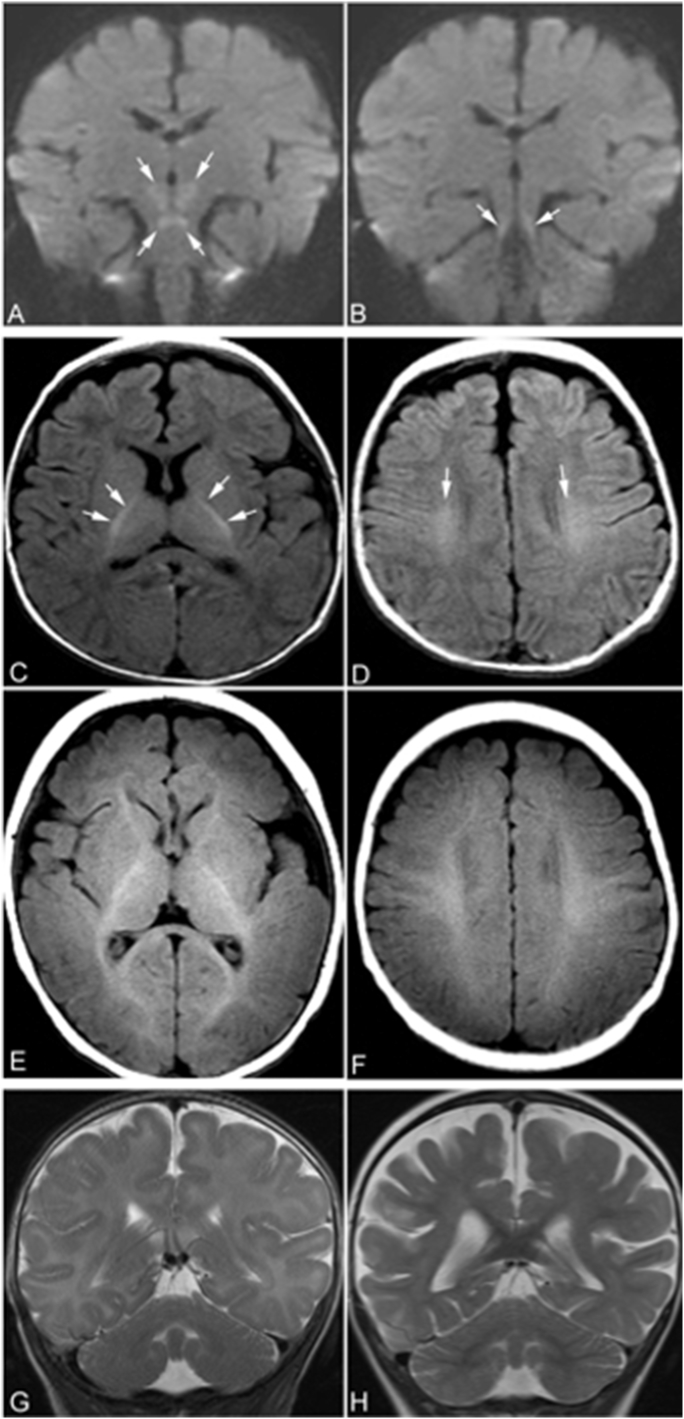
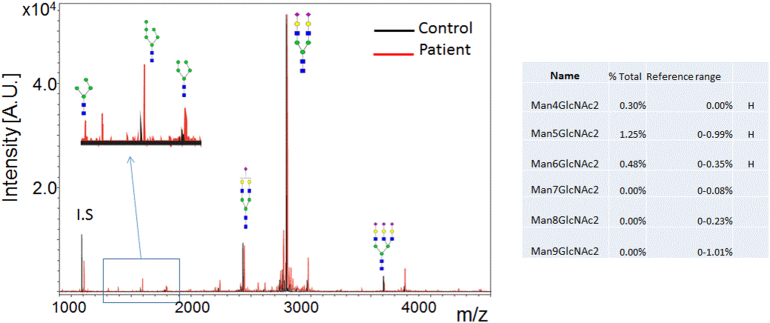
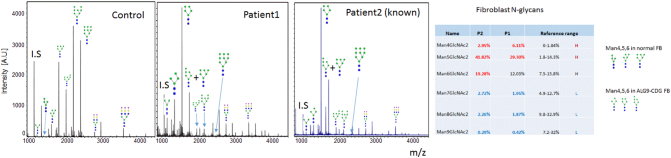
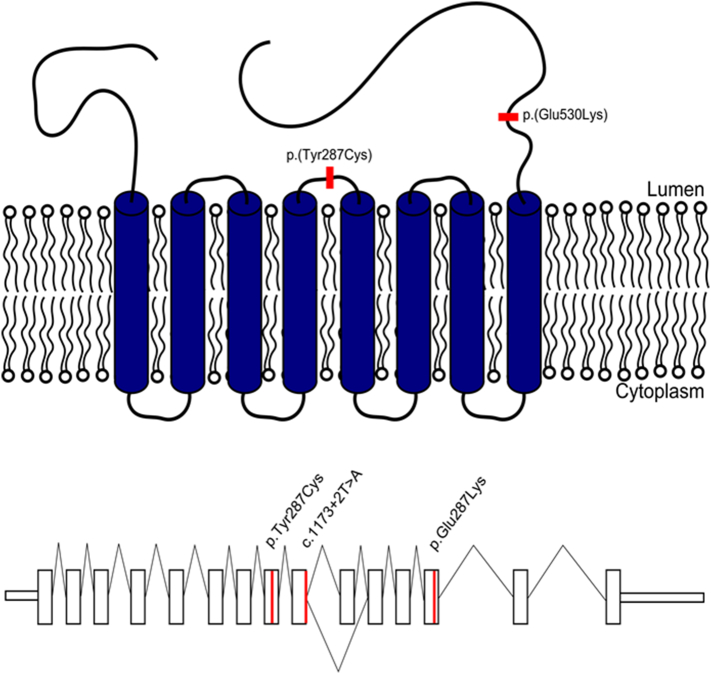
Congenital disorders of glycosylation (CDG) are a group of metabolic diseases resulting from defects in glycan synthesis or processing. The number of subgroups and their phenotypic spectrums continue to expand with most related to deficiencies of N-glycosylation. ALG9-CDG (previously CDG-IL) is the result of a mutation in ALG9. This gene encodes the enzyme alpha-1,2-mannosyltransferase. To date, a total of 10 patients from 6 different families have been reported with one of four ALG9 mutations. Seven of these patients had a similar phenotype with failure to thrive, dysmorphic features, seizures, hepatic and/or renal cysts; the other three patients died in utero from a lethal skeletal dysplasia. This report describes an additional patient with ALG9-CDG who has a milder phenotype. This patient is a term female born to Caucasian, Canadian, non-consanguineous parents of Scottish decent. Prenatally, dysmorphic features, numerous renal cysts and minor cardiac malformations were detected. Post-natally, dysmorphic features included shallow orbits, micrognathia, hypoplastic nipples, talipes equinovarus, lipodystrophy and cutis marmorata. She developed failure to thrive and seizures. The metabolic work-up included analysis of a transferrin isoelectric focusing, which showed a type 1 pattern. This was confirmed by glycan profiling, which identified ahomozygous mutation in ALG9, c.860A > G (p.Tyr287Cys) (NM_1234567890). This had been previously published as a pathogenic mutation in two Canadian patients. Our goal is to contribute to the growing body of knowledge for this disorder by describing the phenotypic spectrum and providing further insight on prognosis.
Keywords: ALG9; ALG9-CDG; CDG-IL; Congenital disorders of glycosylation; Lethal skeletal dysplasia; Transferrin isoelectrofocusing type 1 pattern.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
