

CLIP-seq analysis of multi-mapped reads discovers novel functional RNA regulatory sites in the human transcriptome
- PMID: 28934506
- PMCID: PMC5766199
- DOI: 10.1093/nar/gkx646
CLIP-seq analysis of multi-mapped reads discovers novel functional RNA regulatory sites in the human transcriptome
Abstract
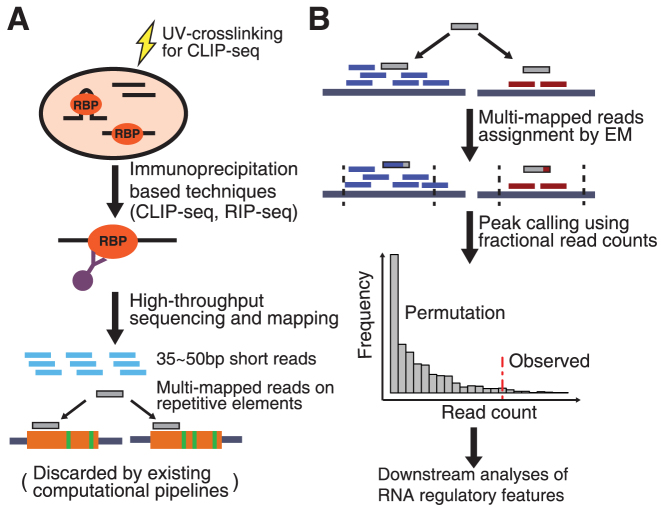
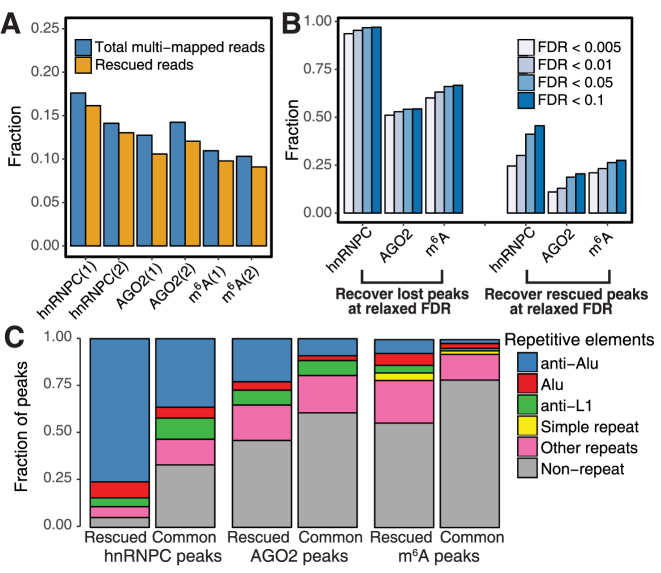
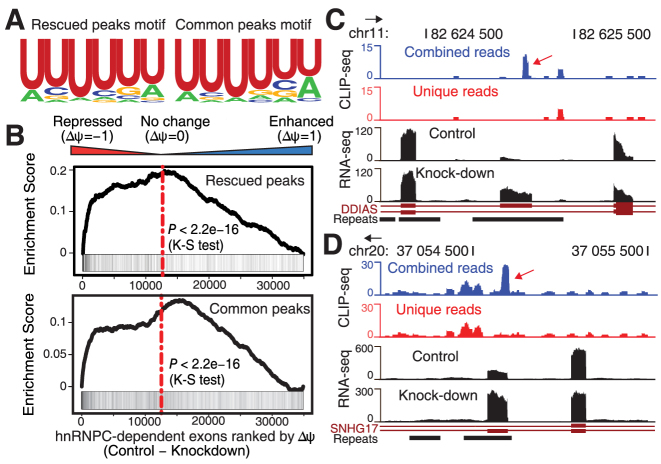
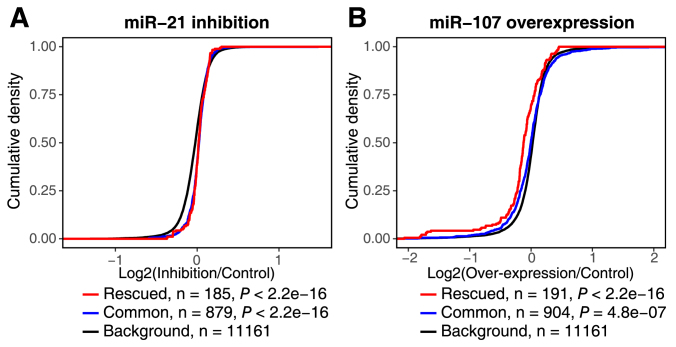
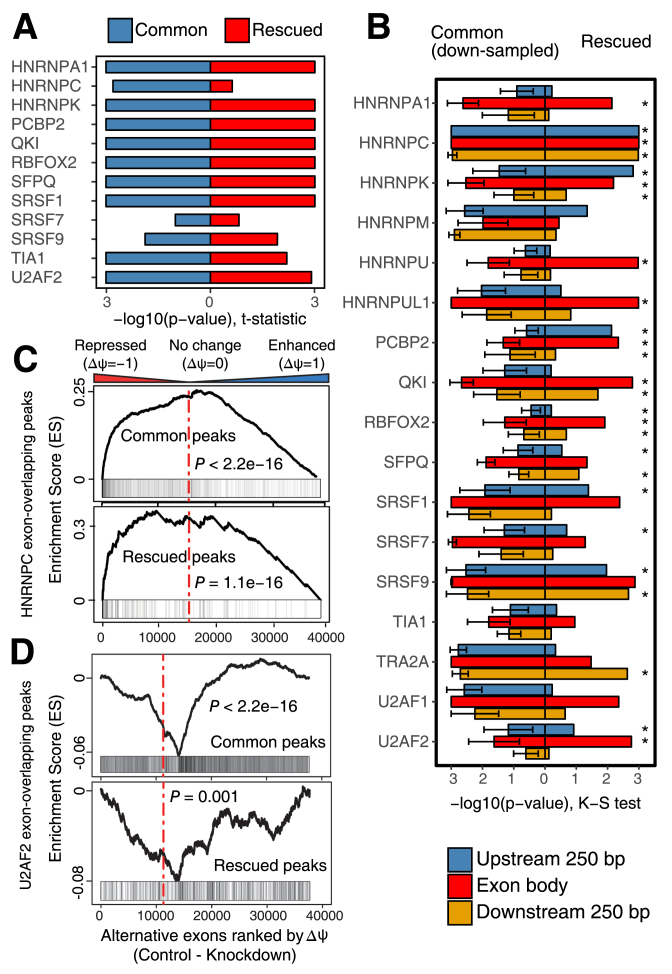
Crosslinking or RNA immunoprecipitation followed by sequencing (CLIP-seq or RIP-seq) allows transcriptome-wide discovery of RNA regulatory sites. As CLIP-seq/RIP-seq reads are short, existing computational tools focus on uniquely mapped reads, while reads mapped to multiple loci are discarded. We present CLAM (CLIP-seq Analysis of Multi-mapped reads). CLAM uses an expectation-maximization algorithm to assign multi-mapped reads and calls peaks combining uniquely and multi-mapped reads. To demonstrate the utility of CLAM, we applied it to a wide range of public CLIP-seq/RIP-seq datasets involving numerous splicing factors, microRNAs and m6A RNA methylation. CLAM recovered a large number of novel RNA regulatory sites inaccessible by uniquely mapped reads. The functional significance of these sites was demonstrated by consensus motif patterns and association with alternative splicing (splicing factors), transcript abundance (AGO2) and mRNA half-life (m6A). CLAM provides a useful tool to discover novel protein-RNA interactions and RNA modification sites from CLIP-seq and RIP-seq data, and reveals the significant contribution of repetitive elements to the RNA regulatory landscape of the human transcriptome.
© The Author(s) 2017. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures

Similar articles
-
Discovering circRNA-microRNA Interactions from CLIP-Seq Data.Methods Mol Biol. 2018;1724:193-207. doi: 10.1007/978-1-4939-7562-4_16. Methods Mol Biol. 2018. PMID: 29322451
-
cCLIP-Seq: Retrieval of Chimeric Reads from HITS-CLIP (CLIP-Seq) Libraries.Methods Mol Biol. 2018;1680:87-100. doi: 10.1007/978-1-4939-7339-2_6. Methods Mol Biol. 2018. PMID: 29030843
-
Computational analysis of CLIP-seq data.Methods. 2017 Apr 15;118-119:60-72. doi: 10.1016/j.ymeth.2017.02.006. Epub 2017 Feb 22. Methods. 2017. PMID: 28254606 Review.
-
A computational approach for identifying microRNA-target interactions using high-throughput CLIP and PAR-CLIP sequencing.BMC Genomics. 2013;14 Suppl 1(Suppl 1):S2. doi: 10.1186/1471-2164-14-S1-S2. Epub 2013 Jan 21. BMC Genomics. 2013. PMID: 23368412 Free PMC article.
-
Recent computational developments on CLIP-seq data analysis and microRNA targeting implications.Brief Bioinform. 2018 Nov 27;19(6):1290-1301. doi: 10.1093/bib/bbx063. Brief Bioinform. 2018. PMID: 28605404 Free PMC article. Review.
Cited by
-
ChiRA: an integrated framework for chimeric read analysis from RNA-RNA interactome and RNA structurome data.Gigascience. 2021 Jan 29;10(2):giaa158. doi: 10.1093/gigascience/giaa158. Gigascience. 2021. PMID: 33511995 Free PMC article.
-
Data Science Issues in Studying Protein-RNA Interactions with CLIP Technologies.Annu Rev Biomed Data Sci. 2018 Jul 20;1(1):235-261. doi: 10.1146/annurev-biodatasci-080917-013525. Annu Rev Biomed Data Sci. 2018. PMID: 37123514 Free PMC article.
-
Nuclear RNA catabolism controls endogenous retroviruses, gene expression asymmetry, and dedifferentiation.Mol Cell. 2023 Dec 7;83(23):4255-4271.e9. doi: 10.1016/j.molcel.2023.10.036. Epub 2023 Nov 22. Mol Cell. 2023. PMID: 37995687 Free PMC article.
-
N6-Methyladenosine in RNA and DNA: An Epitranscriptomic and Epigenetic Player Implicated in Determination of Stem Cell Fate.Stem Cells Int. 2018 Oct 10;2018:3256524. doi: 10.1155/2018/3256524. eCollection 2018. Stem Cells Int. 2018. PMID: 30405719 Free PMC article. Review.
-
Cell type- and factor-specific nonsense-mediated RNA decay.Nucleic Acids Res. 2025 May 10;53(9):gkaf395. doi: 10.1093/nar/gkaf395. Nucleic Acids Res. 2025. PMID: 40366162 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
