

Host-directed therapies for bacterial and viral infections
- PMID: 28935918
- PMCID: PMC7097079
- DOI: 10.1038/nrd.2017.162
Host-directed therapies for bacterial and viral infections
Abstract
Despite the recent increase in the development of antivirals and antibiotics, antimicrobial resistance and the lack of broad-spectrum virus-targeting drugs are still important issues and additional alternative approaches to treat infectious diseases are urgently needed. Host-directed therapy (HDT) is an emerging approach in the field of anti-infectives. The strategy behind HDT is to interfere with host cell factors that are required by a pathogen for replication or persistence, to enhance protective immune responses against a pathogen, to reduce exacerbated inflammation and to balance immune reactivity at sites of pathology. Although HDTs encompassing interferons are well established for the treatment of chronic viral hepatitis, novel strategies aimed at the functional cure of persistent viral infections and the development of broad-spectrum antivirals against emerging viruses seem to be crucial. In chronic bacterial infections, such as tuberculosis, HDT strategies aim to enhance the antimicrobial activities of phagocytes and to curtail inflammation through interference with soluble factors (such as eicosanoids and cytokines) or cellular factors (such as co-stimulatory molecules). This Review describes current progress in the development of HDTs for viral and bacterial infections, including sepsis, and the challenges in bringing these new approaches to the clinic.
Conflict of interest statement
R.S.H. acknowledges research support from Bristol-Myers Squibb and GlaxoSmithKline.
Figures
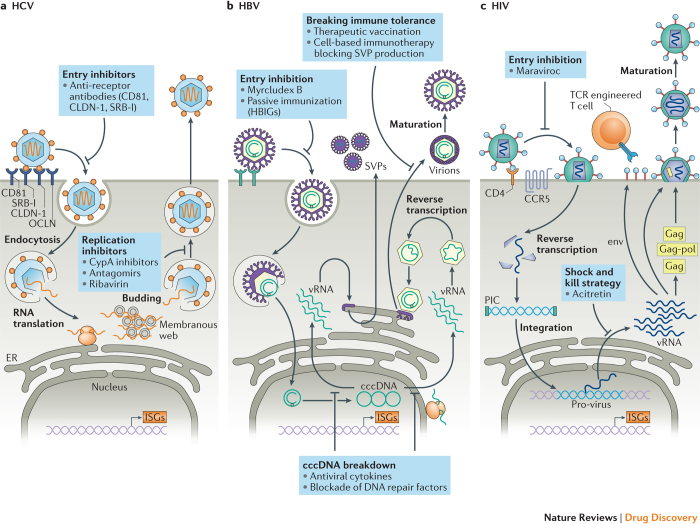
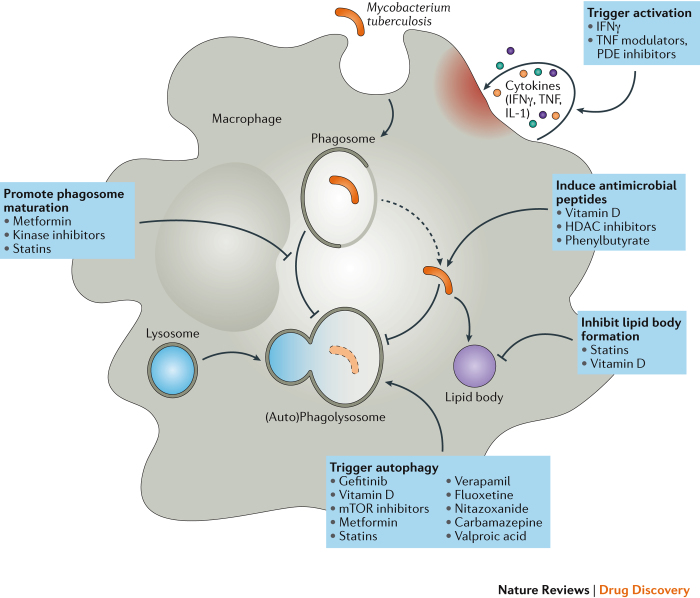
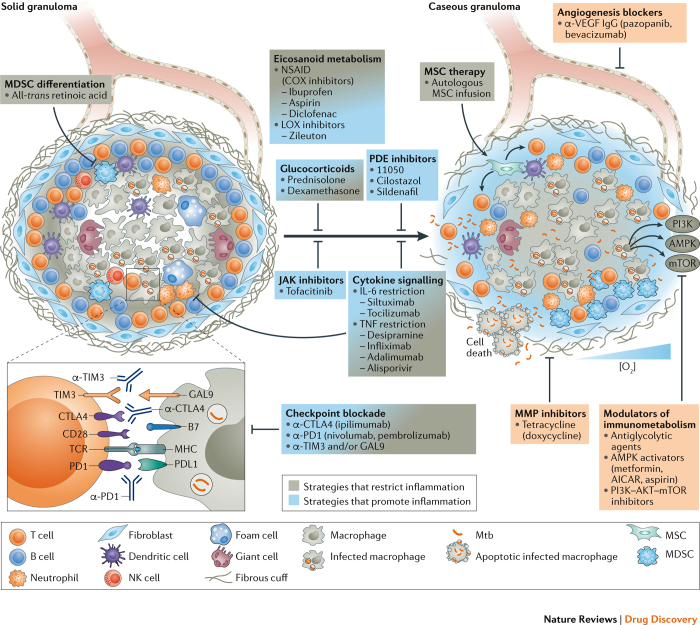
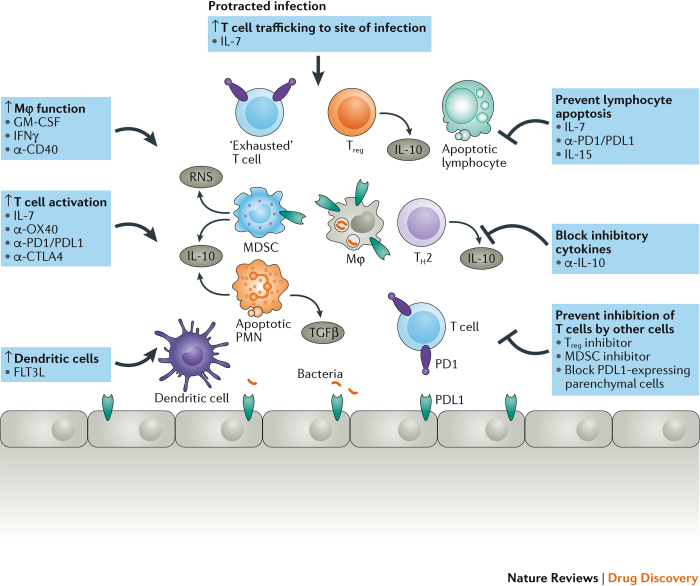
References
-
- O'Neill, J. Tackling drug-resistance infections globally: final report and recommendations. Review on Antimicrobial Resistancehttps://amr-review.org/sites/default/files/160525_Final%20paper_with%20c... (2016).
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
