

Strategies for the etiological therapy of cystic fibrosis
- PMID: 28937684
- PMCID: PMC5635223
- DOI: 10.1038/cdd.2017.126
Strategies for the etiological therapy of cystic fibrosis
Abstract
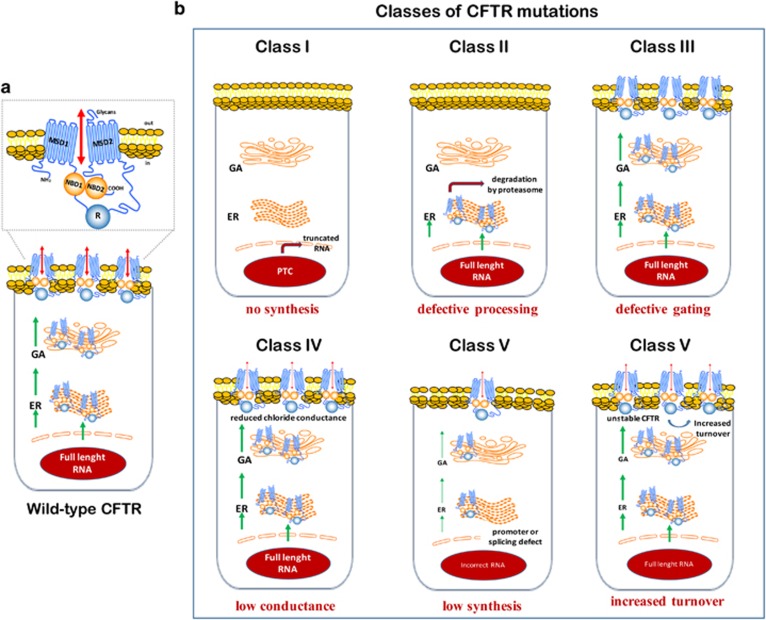
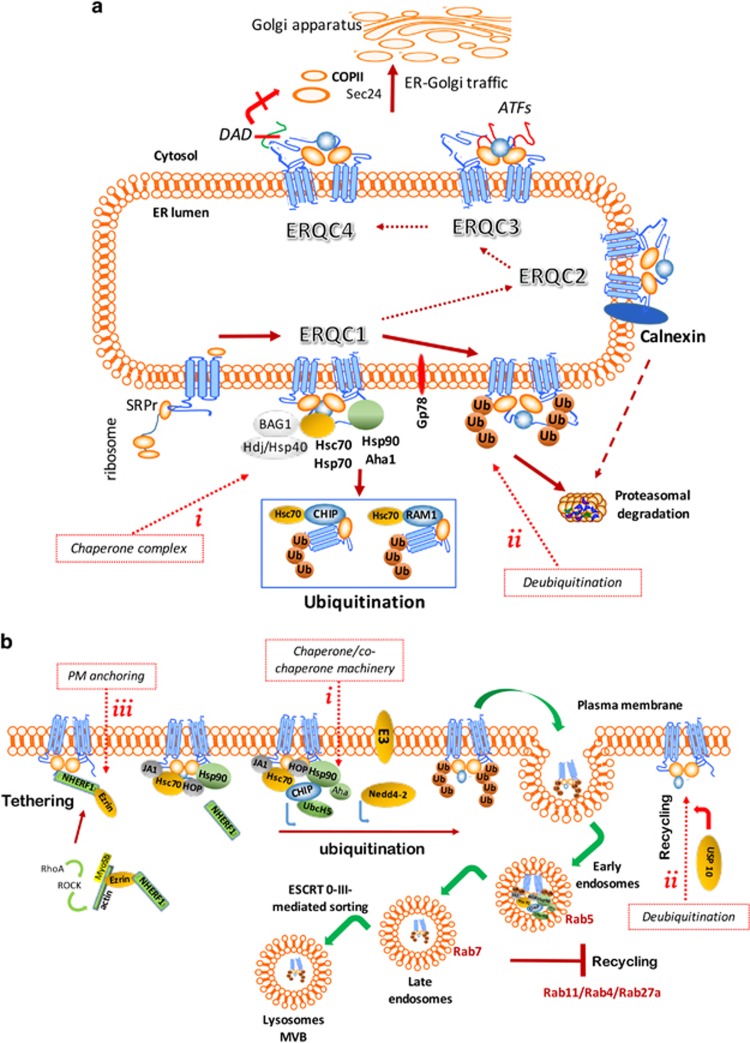
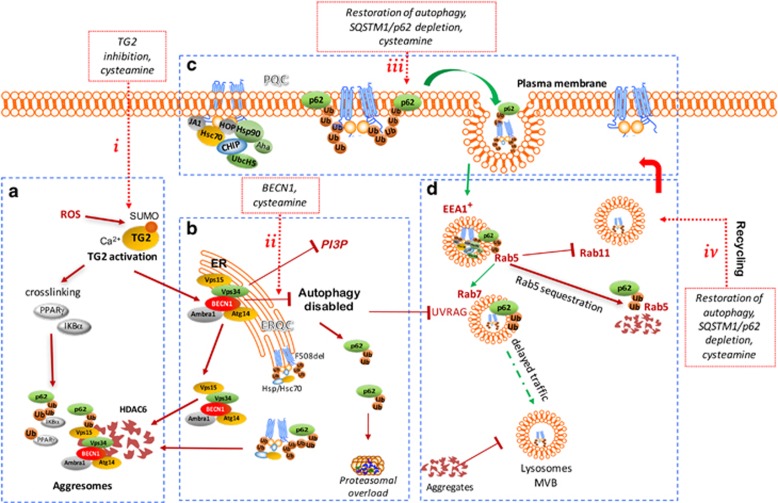
Etiological therapies aim at repairing the underlying cause of cystic fibrosis (CF), which is the functional defect of the cystic fibrosis transmembrane conductance regulator (CFTR) protein owing to mutations in the CFTR gene. Among these, the F508del CFTR mutation accounts for more than two thirds of CF cases worldwide. Two somehow antinomic schools of thought conceive CFTR repair in a different manner. According to one vision, drugs should directly target the mutated CFTR protein to increase its plasma membrane expression (correctors) or improve its ion transport function (potentiators). An alternative strategy consists in modulating the cellular environment and proteostasis networks in which the mutated CFTR protein is synthesized, traffics to its final destination, the plasma membrane, and is turned over. We will analyze distinctive advantages and drawbacks of these strategies in terms of their scientific and clinical dimensions, and we will propose a global strategy for CF research and development based on a reconciliatory approach. Moreover, we will discuss the utility of preclinical biomarkers that may guide the personalized, patient-specific implementation of CF therapies.
Conflict of interest statement
LM, VR and GK are listed as inventors of a patent that protects the use of cysteamine for CF treatment.
Figures
References
-
- Riordan JR. Assembly of functional CFTR chloride channels. Annu Rev Physiol 2005; 67: 701–718. - PubMed
-
- Wang Y, Wrennall JA, Cai Z, Li H, Sheppard DN. Understanding how cystic fibrosis mutations disrupt CFTR function: from single molecules to animal models. Int J Biochem Cell Biol 2014; 52: 47–57. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
