

New Perspectives on Pheochromocytoma and Paraganglioma: Toward a Molecular Classification
- PMID: 28938417
- PMCID: PMC5716829
- DOI: 10.1210/er.2017-00062
New Perspectives on Pheochromocytoma and Paraganglioma: Toward a Molecular Classification
Abstract
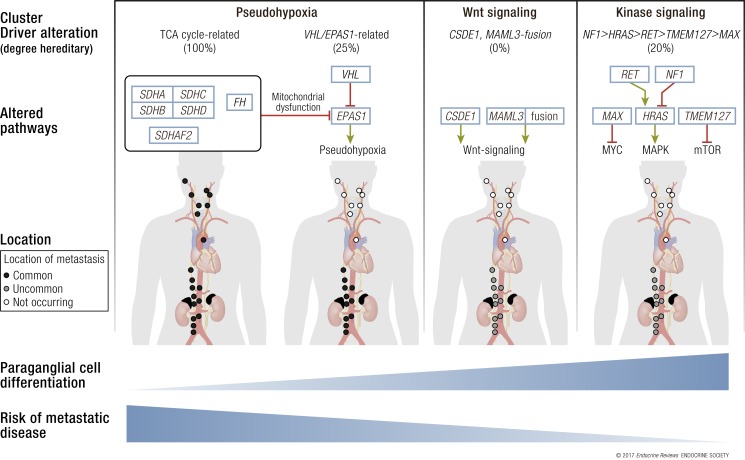
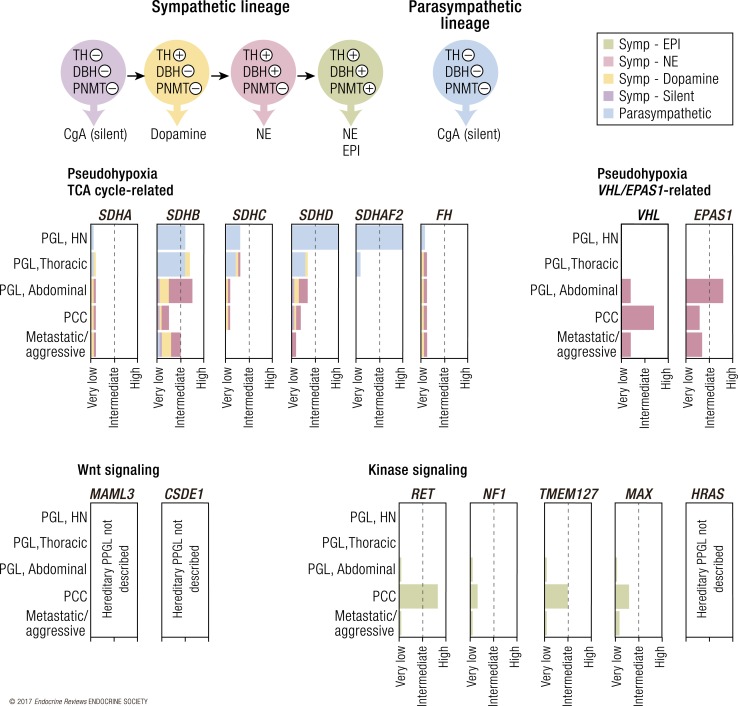
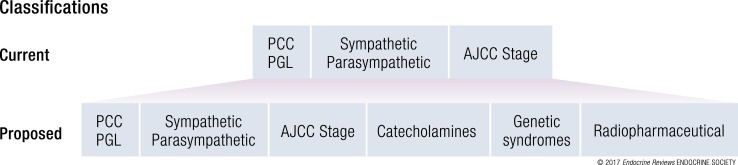
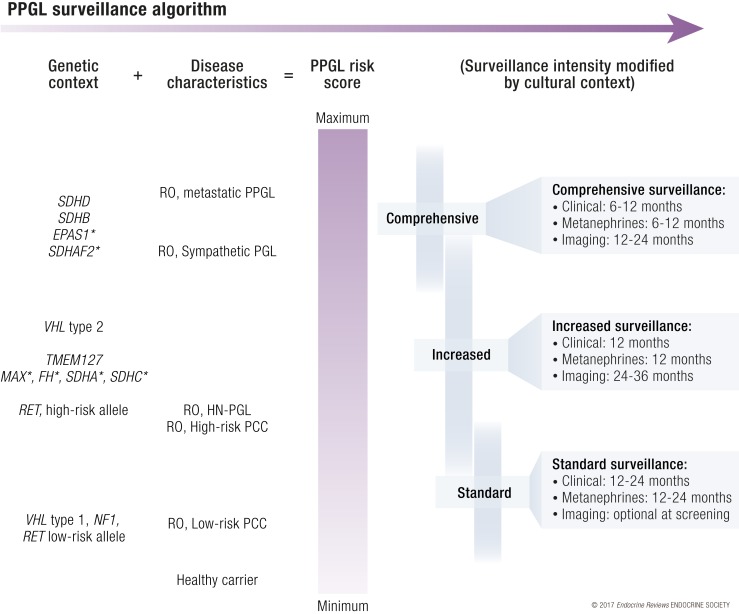
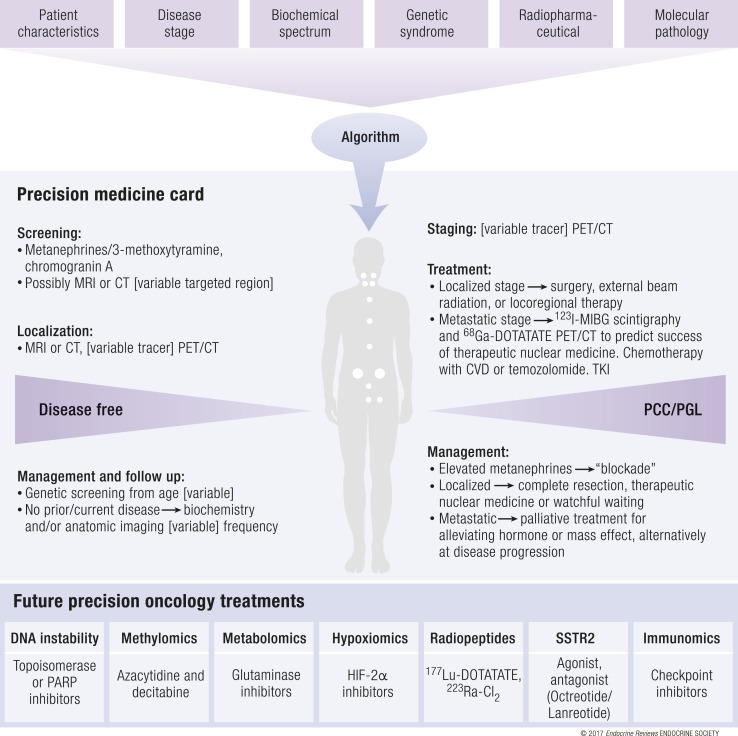
A molecular biology-based taxonomy has been proposed for pheochromocytoma and paraganglioma (PPGL). Data from the Cancer Genome Atlas revealed clinically relevant prognostic and predictive biomarkers and stratified PPGLs into three main clusters. Each subgroup has a distinct molecular-biochemical-imaging signature. Concurrently, new methods for biochemical analysis, functional imaging, and medical therapies have also become available. The research community now strives to match the cluster biomarkers with the best intervention. The concept of precision medicine has been long awaited and holds great promise for improved care. Here, we review the current and future PPGL classifications, with a focus on hereditary syndromes. We discuss the current strengths and shortcomings of precision medicine and suggest a condensed manual for diagnosis and treatment of both adult and pediatric patients with PPGL. Finally, we consider the future direction of this field, with a particular focus on how advanced molecular characterization of PPGL can improve a patient's outcome, including cures and, ultimately, disease prevention.
Copyright © 2017 Endocrine Society.
Figures
References
-
- Lenders JW, Duh QY, Eisenhofer G, Gimenez-Roqueplo AP, Grebe SK, Murad MH, Naruse M, Pacak K, Young WF Jr; Endocrine Society . Pheochromocytoma and paraganglioma: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2014;99(6):1915–1942. - PubMed
-
- Dahia PL. Pheochromocytoma and paraganglioma pathogenesis: learning from genetic heterogeneity. Nat Rev Cancer. 2014;14(2):108–119. - PubMed
-
- Fishbein L, Leshchiner I, Walter V, Danilova L, Robertson AG, Johnson AR, Lichtenberg TM, Murray BA, Ghayee HK, Else T, Ling S, Jefferys SR, de Cubas AA, Wenz B, Korpershoek E, Amelio AL, Makowski L, Rathmell WK, Gimenez-Roqueplo AP, Giordano TJ, Asa SL, Tischler AS, Pacak K, Nathanson KL, Wilkerson MD; Cancer Genome Atlas Research Network . Comprehensive molecular characterization of pheochromocytoma and paraganglioma. Cancer Cell. 2017;31(2):181–193. - PMC - PubMed
-
- Favier J, Amar L, Gimenez-Roqueplo AP. Paraganglioma and phaeochromocytoma: from genetics to personalized medicine. Nat Rev Endocrinol. 2015;11(2):101–111. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
