

Positive transcription elongation factor b (P-TEFb) is a therapeutic target in human multiple myeloma
- PMID: 28938651
- PMCID: PMC5601747
- DOI: 10.18632/oncotarget.19761
Positive transcription elongation factor b (P-TEFb) is a therapeutic target in human multiple myeloma
Abstract
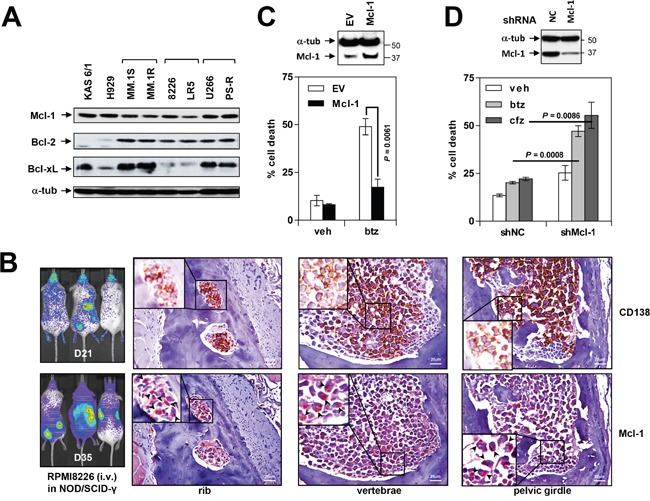
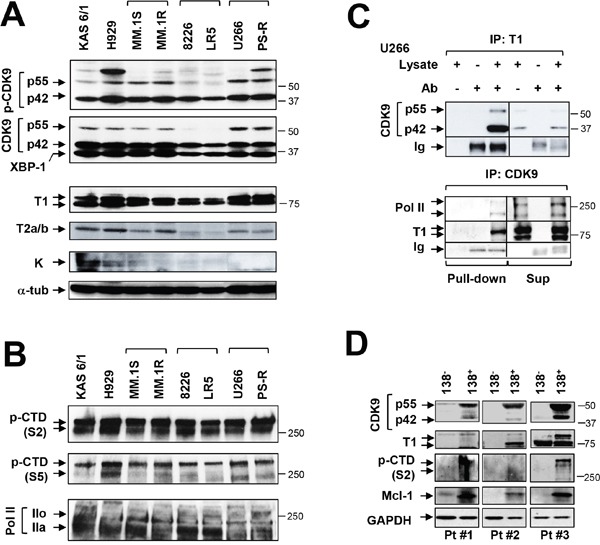
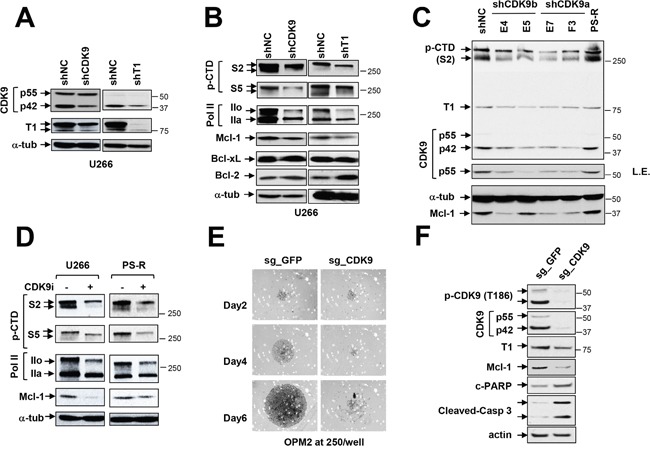
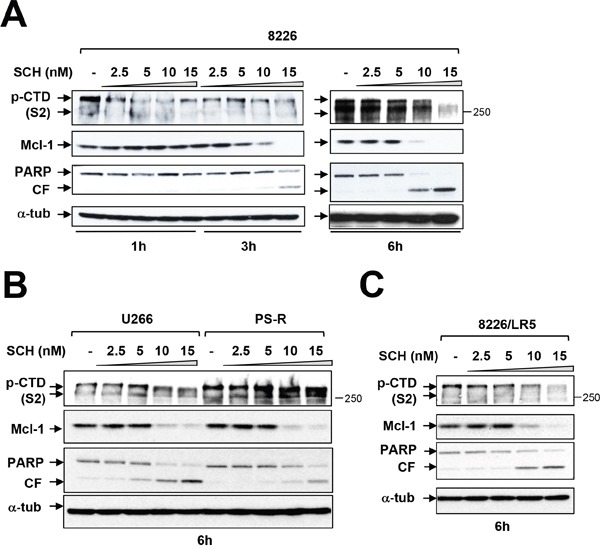
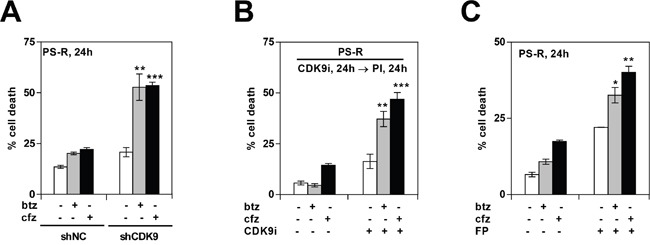
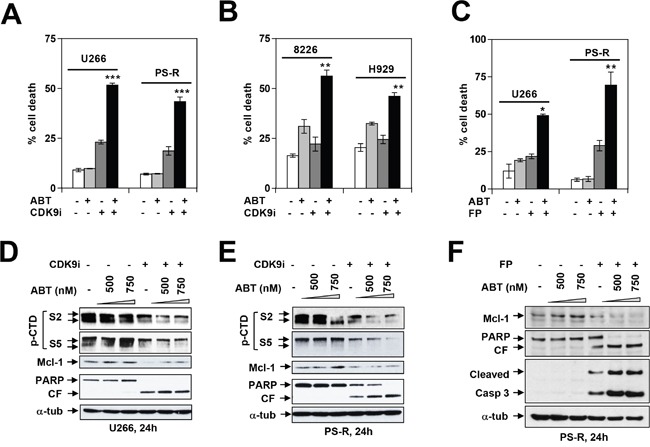
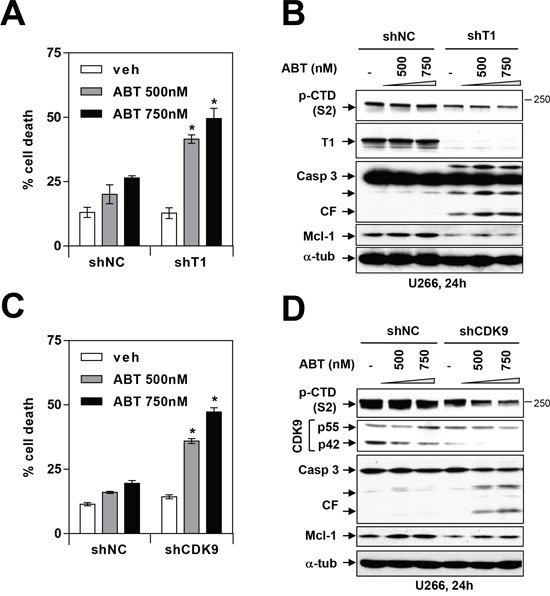
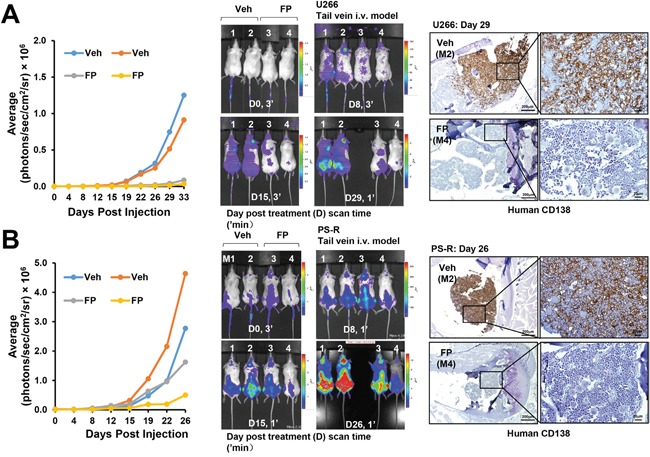
The role of the positive RNA Pol II regulator, P-TEFb (positive transcription elongation factor b), in maintenance of the anti-apoptotic protein Mcl-1 and bortezomib (btz) resistance was investigated in human multiple myeloma (MM) cells. Mcl-1 was up-regulated in all MM lines tested, including bortezomib-resistant lines, human MM xenograft mouse models, and primary CD138+ MM cells. Mcl-1 over-expression significantly reduced bortezomib lethality, indicating a functional role for Mcl-1 in bortezomib resistance. MM cell lines, primary MM specimens, and murine xenografts exhibited constitutive P-TEFb activation, manifested by high CTD (carboxy-terminal domain) S2 phosphorylation, associated with a) P-TEFb subunit up-regulation i.e., CDK9 (42 and 55 kDa isoforms) and cyclin T1; and b) marked CDK9 (42 kDa) T186 phosphorylation. In marked contrast, normal hematopoietic cells failed to exhibit up-regulation of p-CTD, CDK9, cyclin T1, or Mcl-1. CDK9 or cyclin T1 shRNA knock-down dramatically inhibited CTD S2 phosphorylation and down-regulated Mcl-1. Moreover, CRISPR-Cas CDK9 knock-out triggered apoptosis in MM cells and dramatically diminished cell growth. Pan-CDK e.g., dinaciclib or alvocidib and selective CDK9 inhibitors (CDK9i) recapitulated the effects of genetic P-TEFb disruption. CDK9 shRNA or CDK9 inhibitors significantly potentiated the susceptibility of MM cells, including bortezomib-resistant cells, to proteasome inhibitors. Analogously, CDK9 or cyclin T1 knock-down or CDK9 inhibitors markedly increased BH3-mimetic lethality in bortezomib-resistant cells. Finally, pan-CDK inhibition reduced human drug-naïve or bortezomib-resistant CD138+ cells and restored bone marrow architecture in vivo. Collectively, these findings implicate constitutive P-TEFb activation in high Mcl-1 maintenance in MM, and validate targeting the P-TEFb complex to circumvent bortezomib-resistance.
Keywords: CDK inhibitors; MCL-1; P-TEFb; bortezomib resistance; myeloma.
Conflict of interest statement
CONFLICTS OF INTEREST No potential conflicts of interest were disclosed.
Figures
Similar articles
-
Bromodomain protein Brd4 regulates human immunodeficiency virus transcription through phosphorylation of CDK9 at threonine 29.J Virol. 2009 Jan;83(2):1036-44. doi: 10.1128/JVI.01316-08. Epub 2008 Oct 29. J Virol. 2009. PMID: 18971272 Free PMC article.
-
Transcription Elongation Factor P-TEFb Is Involved in IL-17F Signaling in Airway Smooth Muscle Cells.Int Arch Allergy Immunol. 2018;176(2):83-90. doi: 10.1159/000488154. Epub 2018 Apr 12. Int Arch Allergy Immunol. 2018. PMID: 29649811
-
Peroxisome proliferator-activated receptor gamma recruits the positive transcription elongation factor b complex to activate transcription and promote adipogenesis.Mol Endocrinol. 2006 Jul;20(7):1494-505. doi: 10.1210/me.2005-0222. Epub 2006 Feb 16. Mol Endocrinol. 2006. PMID: 16484339 Free PMC article.
-
The emerging picture of CDK9/P-TEFb: more than 20 years of advances since PITALRE.Mol Biosyst. 2017 Jan 31;13(2):246-276. doi: 10.1039/c6mb00387g. Mol Biosyst. 2017. PMID: 27833949 Review.
-
Targeting CDK9 for Anti-Cancer Therapeutics.Cancers (Basel). 2021 May 1;13(9):2181. doi: 10.3390/cancers13092181. Cancers (Basel). 2021. PMID: 34062779 Free PMC article. Review.
Cited by
-
An Omics Approach to Extracellular Vesicles from HIV-1 Infected Cells.Cells. 2019 Jul 29;8(8):787. doi: 10.3390/cells8080787. Cells. 2019. PMID: 31362387 Free PMC article.
-
Multiple myeloma: Combination therapy of BET proteolysis targeting chimeric molecule with CDK9 inhibitor.PLoS One. 2020 Jun 19;15(6):e0232068. doi: 10.1371/journal.pone.0232068. eCollection 2020. PLoS One. 2020. PMID: 32559187 Free PMC article.
-
Cyclin-dependent kinase 9 as a potential specific molecular target in NK-cell leukemia/lymphoma.Haematologica. 2018 Dec;103(12):2059-2068. doi: 10.3324/haematol.2018.191395. Epub 2018 Aug 3. Haematologica. 2018. PMID: 30076184 Free PMC article.
-
Peptidyl Arginine Deiminase 2 (PADI2)-Mediated Arginine Citrullination Modulates Transcription in Cancer.Int J Mol Sci. 2020 Feb 17;21(4):1351. doi: 10.3390/ijms21041351. Int J Mol Sci. 2020. PMID: 32079300 Free PMC article. Review.
-
Genome Editing Approaches with CRISPR/Cas9 for Cancer Treatment: Critical Appraisal of Preclinical and Clinical Utility, Challenges, and Future Research.Cells. 2022 Sep 6;11(18):2781. doi: 10.3390/cells11182781. Cells. 2022. PMID: 36139356 Free PMC article. Review.
References
-
- Wuilleme-Toumi S, Robillard N, Gomez P, Moreau P, Le Gouill S, Avet-Loiseau H, Harousseau JL, Amiot M, Bataille R. Mcl-1 is overexpressed in multiple myeloma and associated with relapse and shorter survival. Leukemia. 2005;19:1248–1252. - PubMed
-
- Derenne S, Monia B, Dean NM, Taylor JK, Rapp MJ, Harousseau JL, Bataille R, Amiot M. Antisense strategy shows that Mcl-1 rather than Bcl-2 or Bcl-x(L) is an essential survival protein of human myeloma cells. Blood. 2002;100:194–199. - PubMed
-
- Zhang B, Gojo I, Fenton RG. Myeloid cell factor-1 is a critical survival factor for multiple myeloma. Blood. 2002;99:1885–1893. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
