

PD-1 Blockade Promotes Epitope Spreading in Anticancer CD8+ T Cell Responses by Preventing Fratricidal Death of Subdominant Clones To Relieve Immunodomination
- PMID: 28939757
- PMCID: PMC5731479
- DOI: 10.4049/jimmunol.1700643
PD-1 Blockade Promotes Epitope Spreading in Anticancer CD8+ T Cell Responses by Preventing Fratricidal Death of Subdominant Clones To Relieve Immunodomination
Abstract
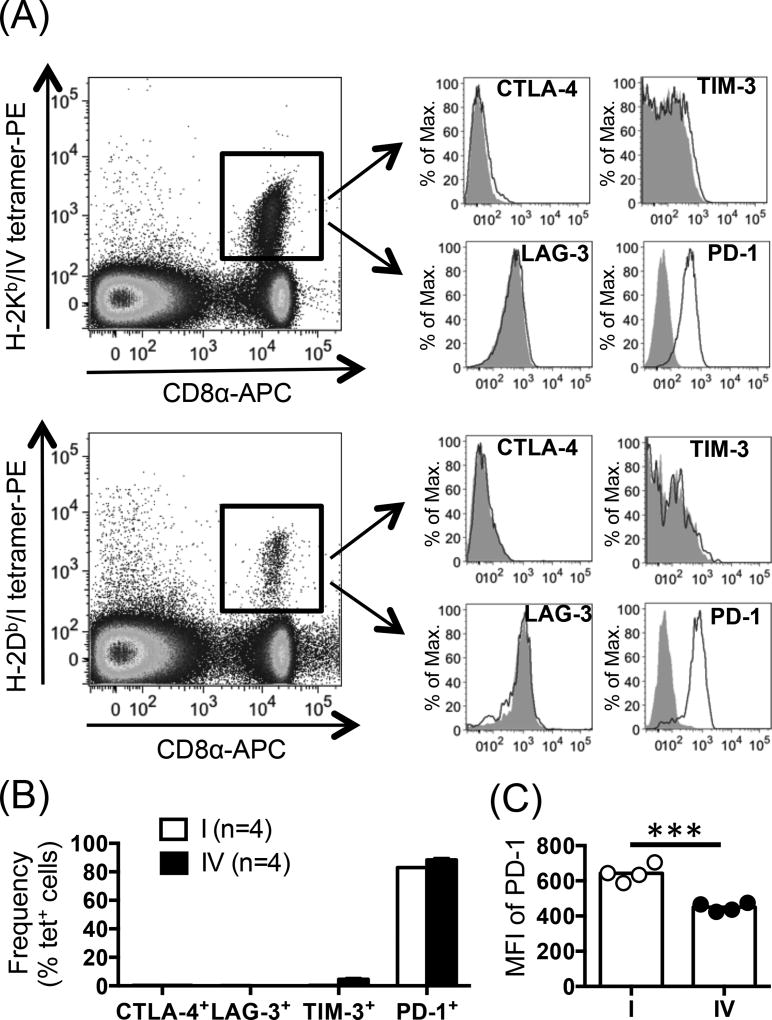
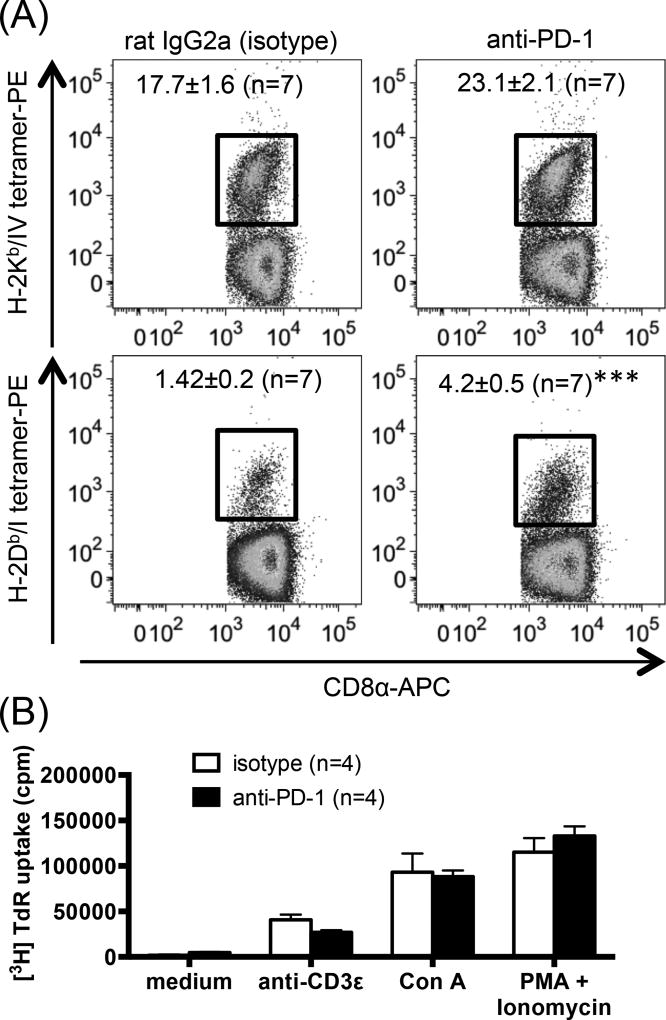
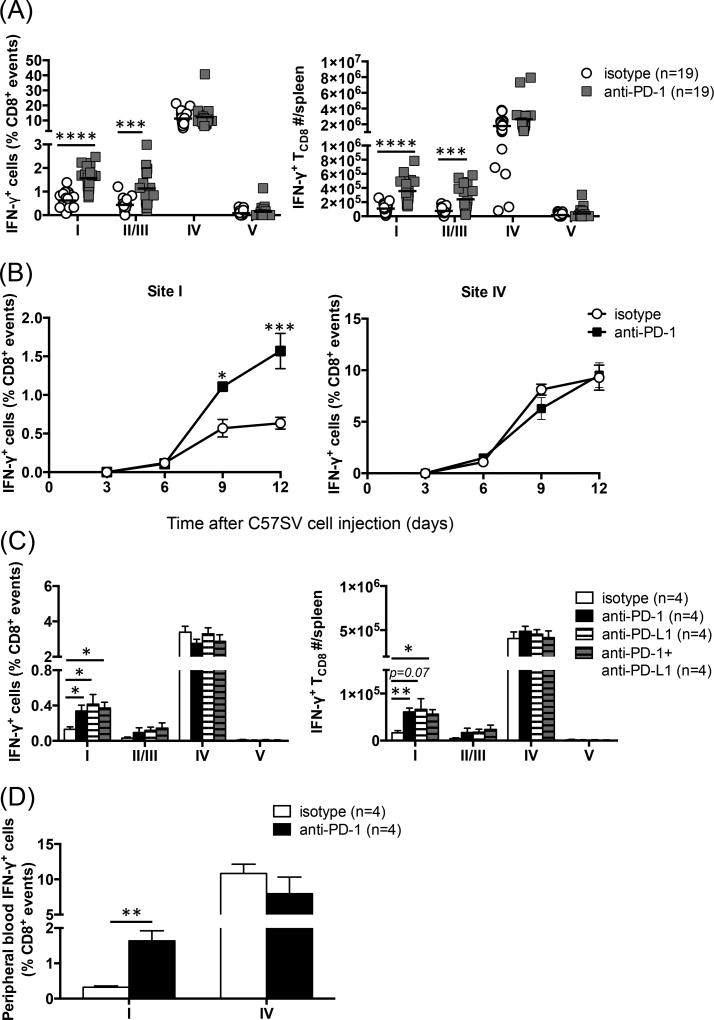
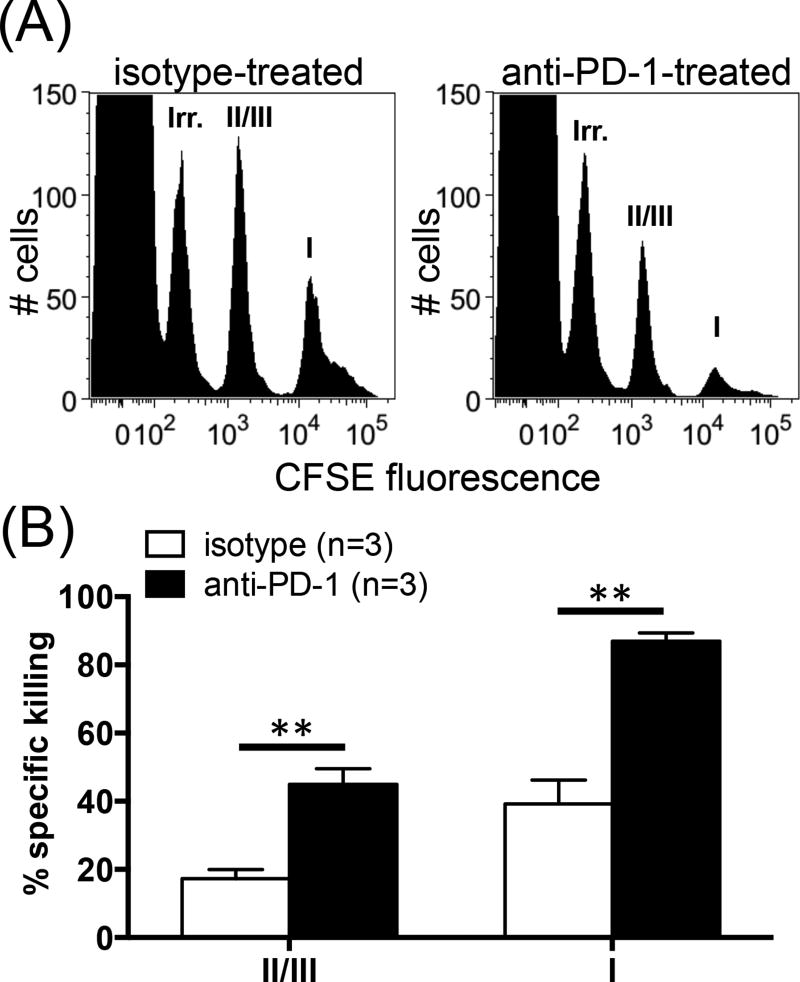
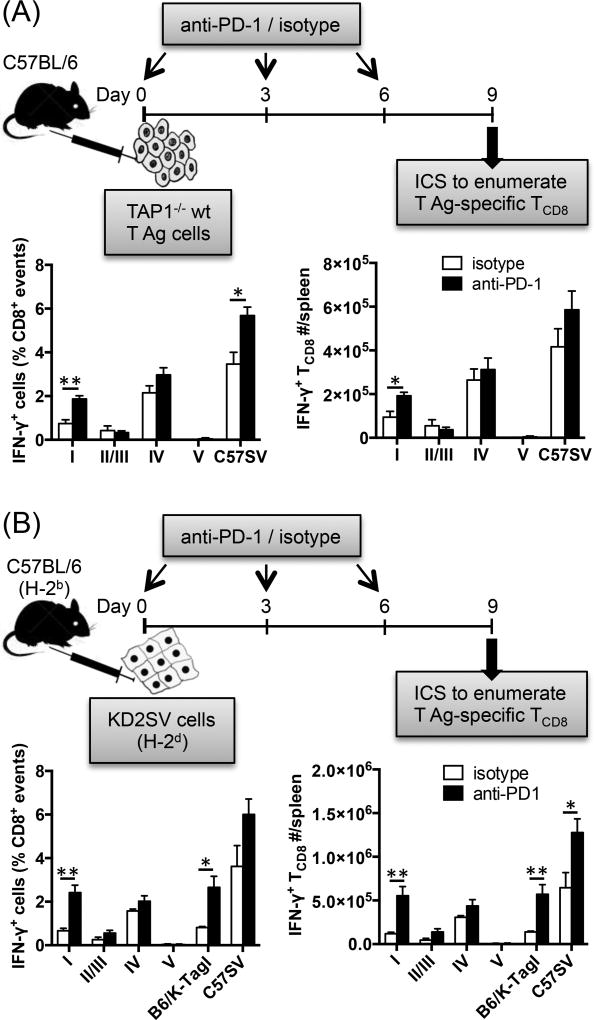
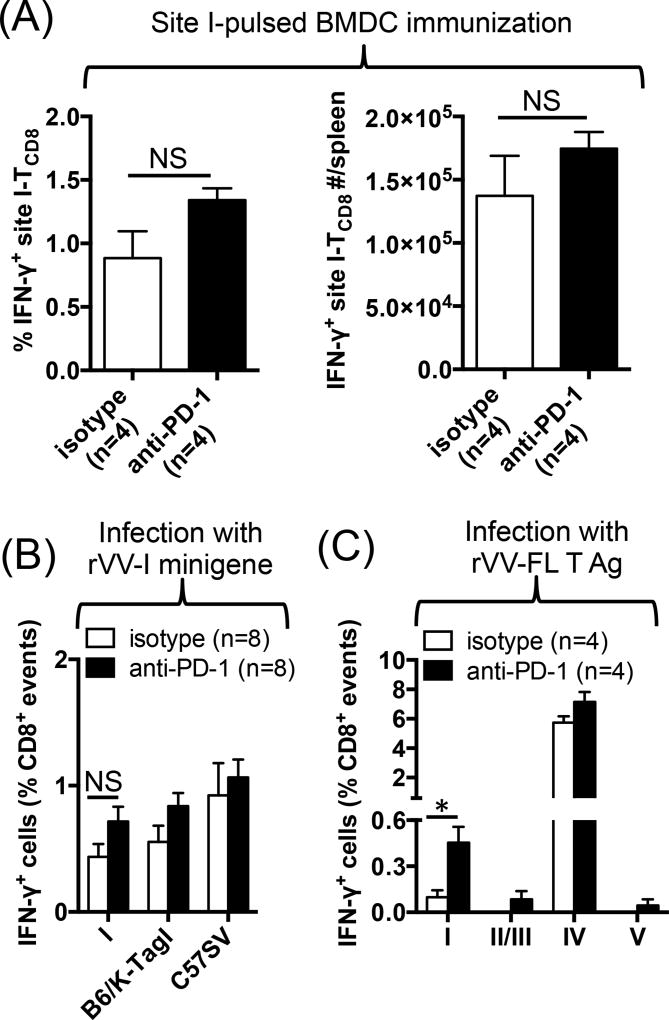
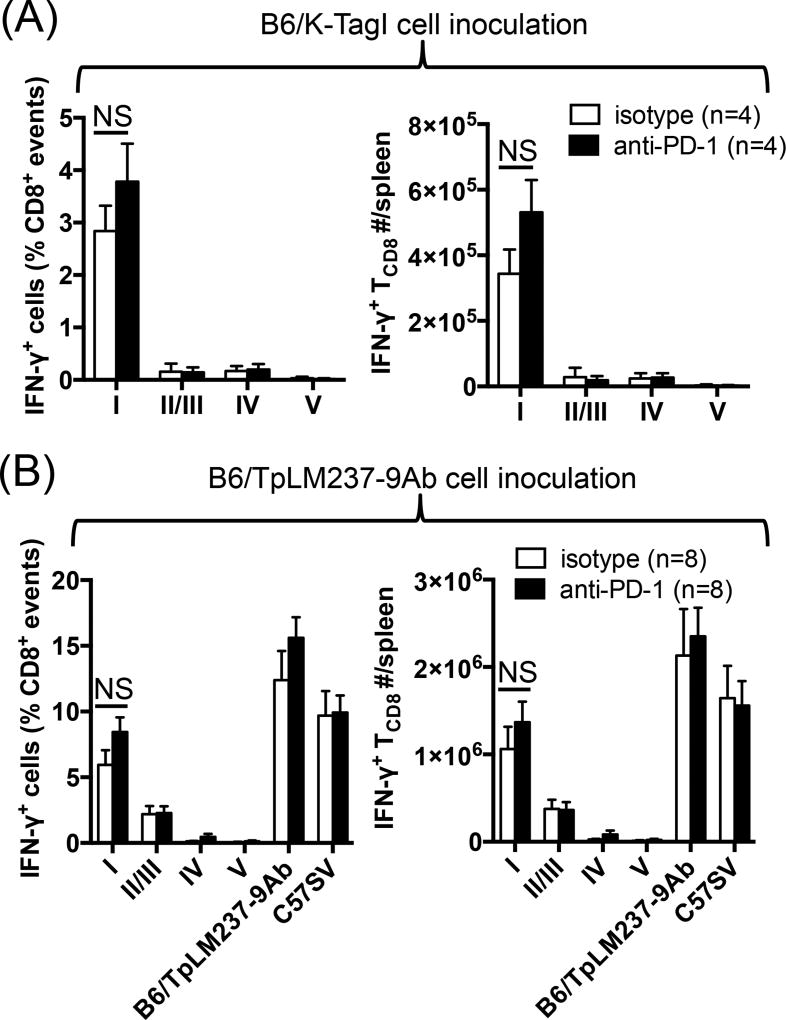
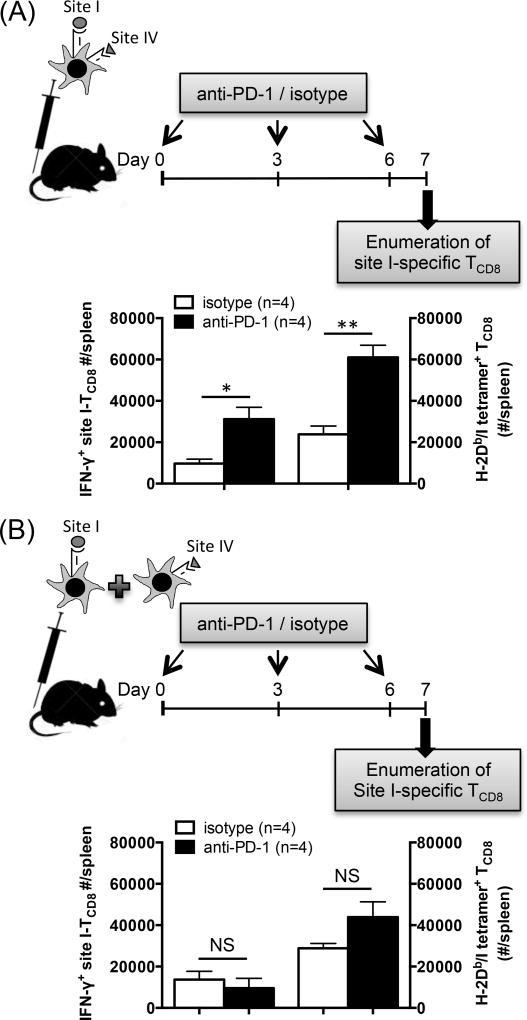
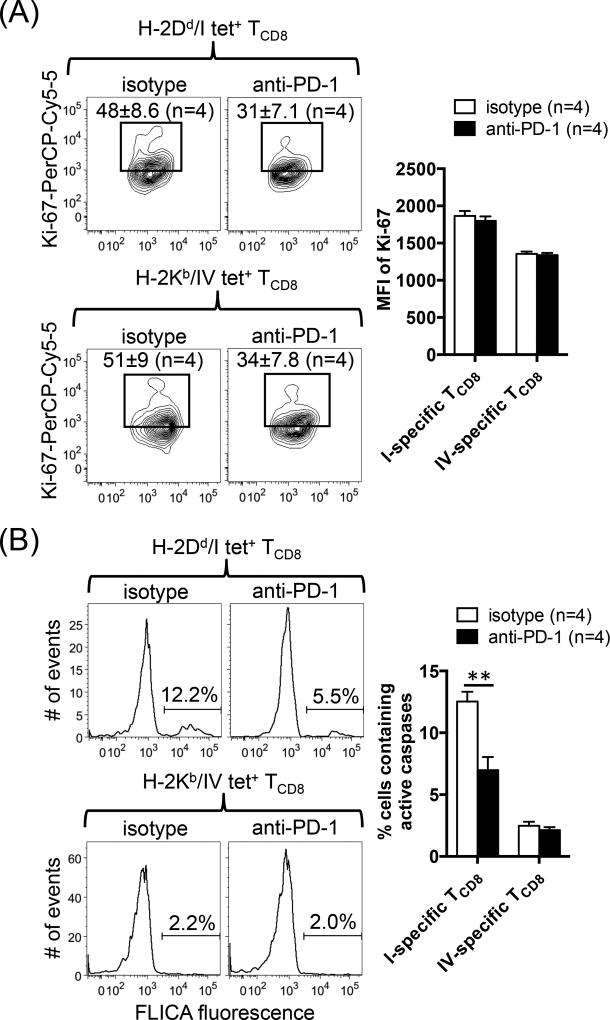
The interactions between programmed death-1 (PD-1) and its ligands hamper tumor-specific CD8+ T cell (TCD8) responses, and PD-1-based "checkpoint inhibitors" have shown promise in certain cancers, thus revitalizing interest in immunotherapy. PD-1-targeted therapies reverse TCD8 exhaustion/anergy. However, whether they alter the epitope breadth of TCD8 responses remains unclear. This is an important question because subdominant TCD8 are more likely than immunodominant clones to escape tolerance mechanisms and may contribute to protective anticancer immunity. We have addressed this question in an in vivo model of TCD8 responses to well-defined epitopes of a clinically relevant oncoprotein, large T Ag. We found that unlike other coinhibitory molecules (CTLA-4, LAG-3, TIM-3), PD-1 was highly expressed by subdominant TCD8, which correlated with their propensity to favorably respond to PD-1/PD-1 ligand-1 (PD-L1)-blocking Abs. PD-1 blockade increased the size of subdominant TCD8 clones at the peak of their primary response, and it also sustained their presence, thus giving rise to an enlarged memory pool. The expanded population was fully functional as judged by IFN-γ production and MHC class I-restricted cytotoxicity. The selective increase in subdominant TCD8 clonal size was due to their enhanced survival, not proliferation. Further mechanistic studies utilizing peptide-pulsed dendritic cells, recombinant vaccinia viruses encoding full-length T Ag or epitope mingenes, and tumor cells expressing T Ag variants revealed that anti-PD-1 invigorates subdominant TCD8 responses by relieving their lysis-dependent suppression by immunodominant TCD8 To our knowledge, our work constitutes the first report that interfering with PD-1 signaling potentiates epitope spreading in tumor-specific responses, a finding with clear implications for cancer immunotherapy and vaccination.
Copyright © 2017 by The American Association of Immunologists, Inc.
Figures
References
-
- Gallimore A, Glithero A, Godkin A, Tissot AC, Pluckthun A, Elliott T, Hengartner H, Zinkernagel R. Induction and exhaustion of lymphocytic choriomeningitis virus-specific cytotoxic T lymphocytes visualized using soluble tetrameric major histocompatibility complex class I-peptide complexes. J Exp Med. 1998;187:1383–1393. - PMC - PubMed
-
- Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348:56–61. - PubMed
-
- Hoffmann M, Pantazis N, Martin GE, Hickling S, Hurst J, Meyerowitz J, Willberg CB, Robinson N, Brown H, Fisher M, Kinloch S, Babiker A, Weber J, Nwokolo N, Fox J, Fidler S, Phillips R, Frater J Spartac, and C. Investigators. Exhaustion of Activated CD8 T Cells Predicts Disease Progression in Primary HIV-1 Infection. PLoS Pathog. 2016;12:e1005661. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
