

Sodium Channelopathies of Skeletal Muscle
- PMID: 28939973
- PMCID: PMC5866235
- DOI: 10.1007/164_2017_52
Sodium Channelopathies of Skeletal Muscle
Abstract
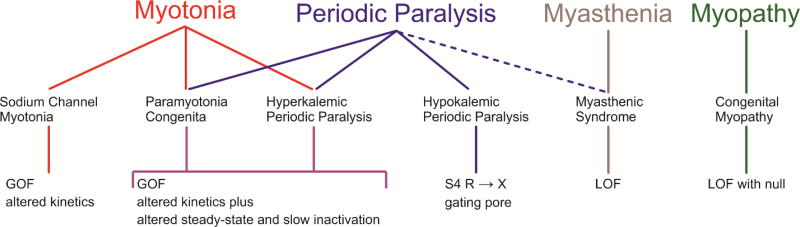
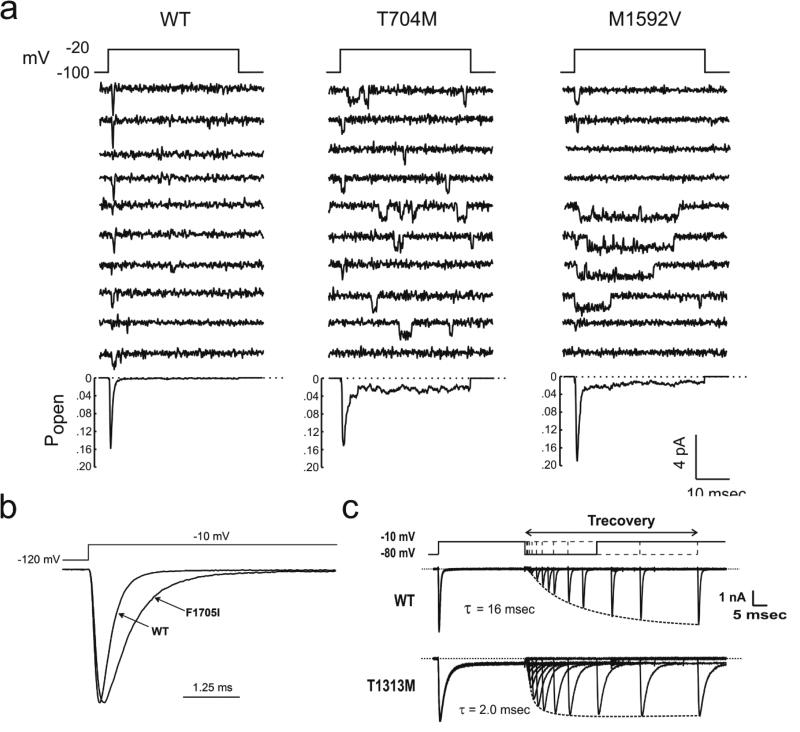
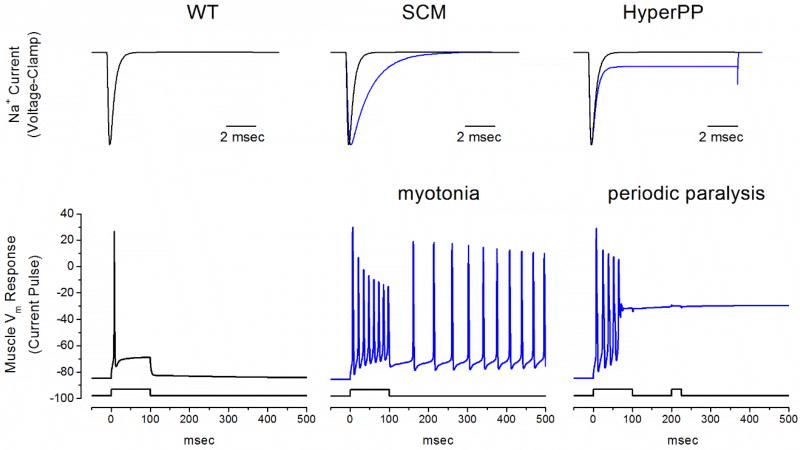
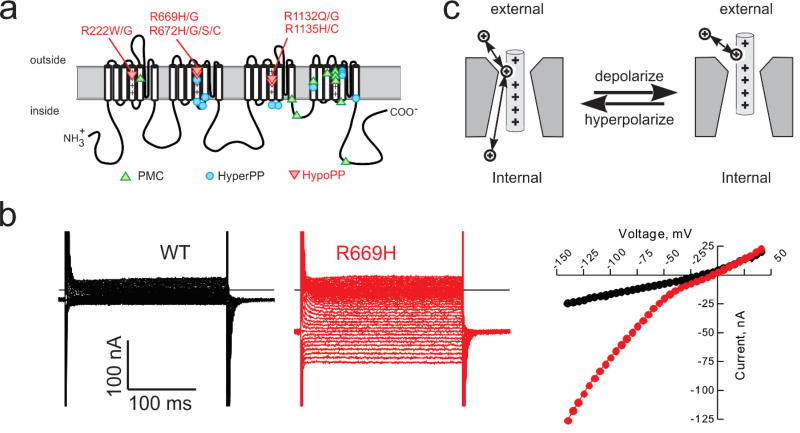
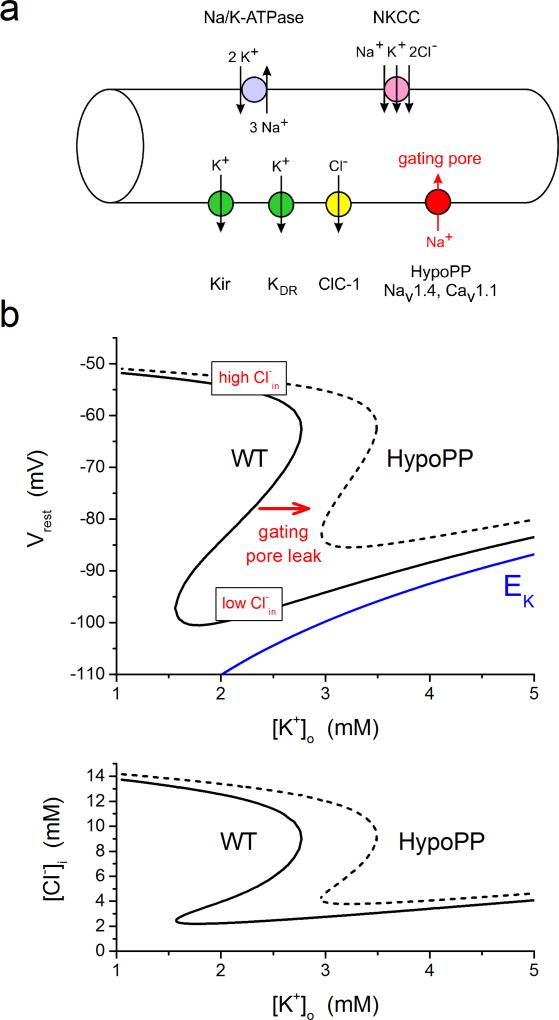
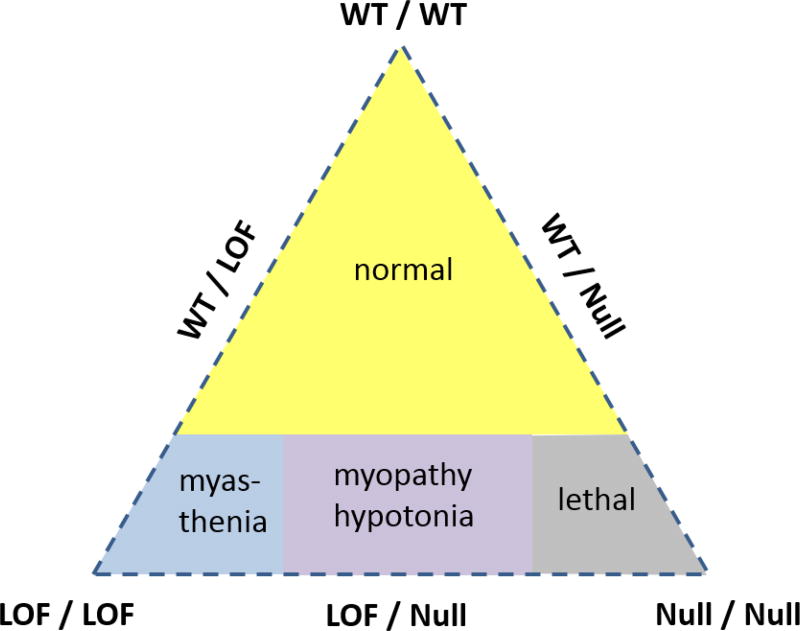
The NaV1.4 sodium channel is highly expressed in skeletal muscle, where it carries almost all of the inward Na+ current that generates the action potential, but is not present at significant levels in other tissues. Consequently, mutations of SCN4A encoding NaV1.4 produce pure skeletal muscle phenotypes that now include six allelic disorders: sodium channel myotonia, paramyotonia congenita, hyperkalemic periodic paralysis, hypokalemic periodic paralysis, congenital myasthenia, and congenital myopathy with hypotonia. Mutation-specific alternations of NaV1.4 function explain the mechanistic basis for the diverse phenotypes and identify opportunities for strategic intervention to modify the burden of disease.
Keywords: Channelopathy; Gating pore; Myotonia; NaV1.4; Periodic paralysis; Sodium channel.
Figures
References
-
- Brancati F, Valente EM, Davies NP, Sarkozy A, Sweeney MG, LoMonaco M, Pizzuti A, Hanna MG, Dallapiccola B. Severe infantile hyperkalaemic periodic paralysis and paramyotonia congenita: broadening the clinical spectrum associated with the T704M mutation in SCN4A. J Neurol Neurosurg Psychiatry. 2003;74:1339–1341. - PMC - PubMed
-
- Brown GL, Harvey AM. Congenital myotonia in the goat. Brain. 1939;62:341–363.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
