

Mitochondrial Fission Promotes the Continued Clearance of Apoptotic Cells by Macrophages
- PMID: 28942921
- PMCID: PMC5679712
- DOI: 10.1016/j.cell.2017.08.041
Mitochondrial Fission Promotes the Continued Clearance of Apoptotic Cells by Macrophages
Abstract
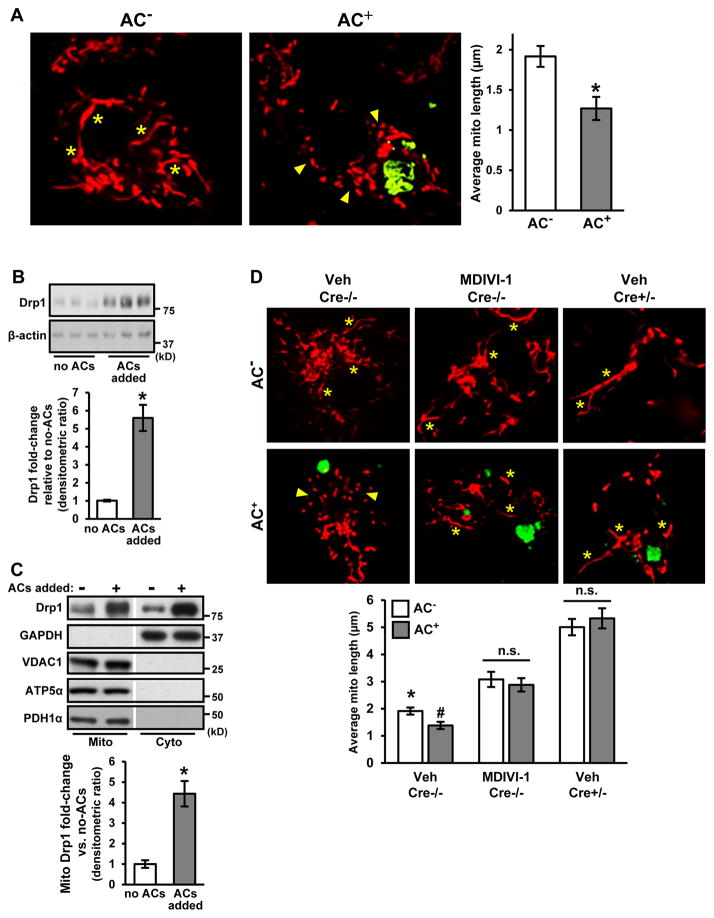
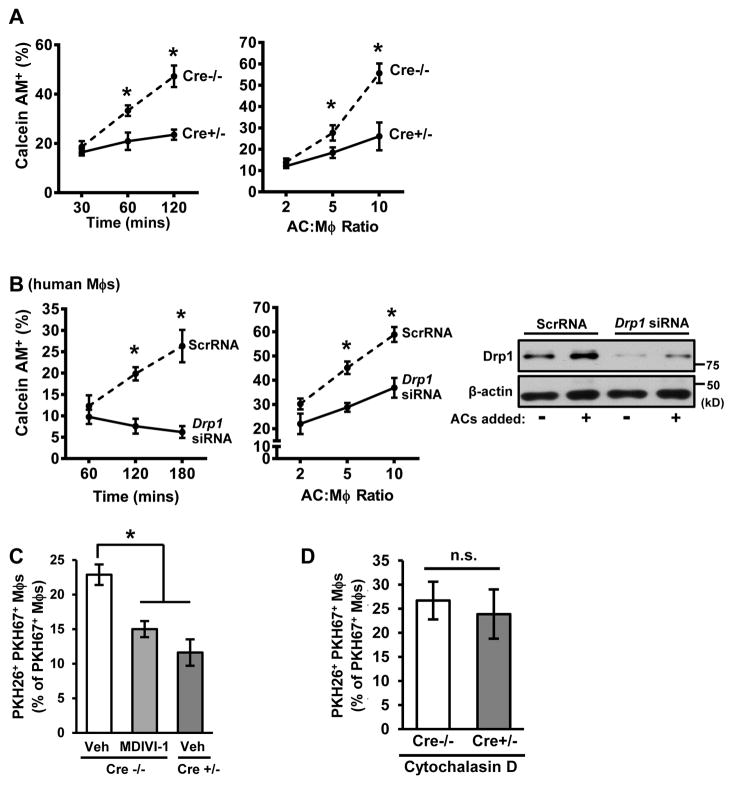
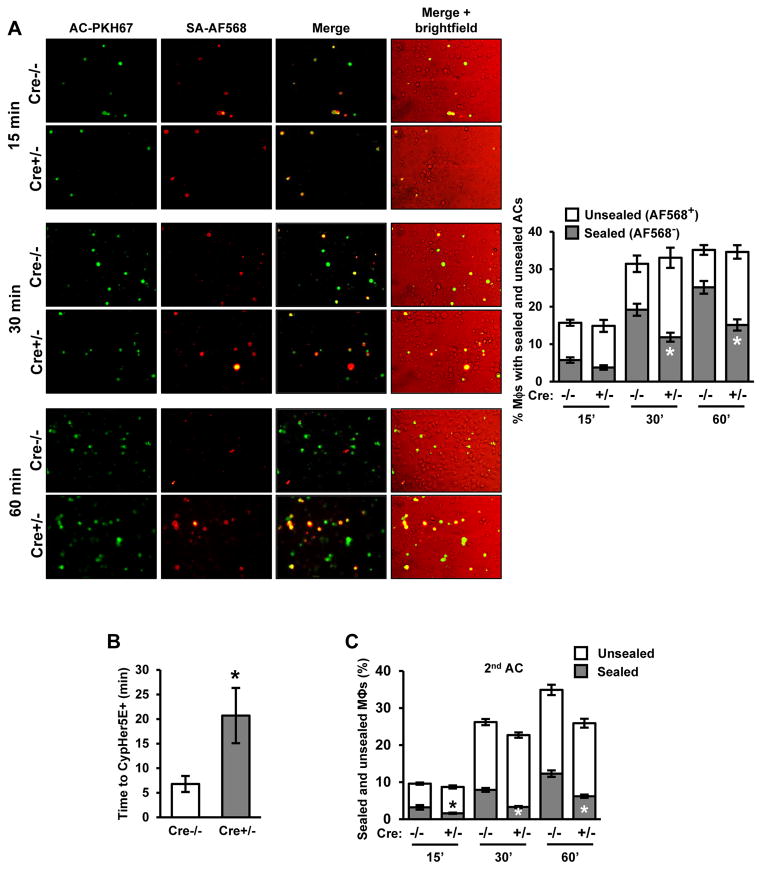
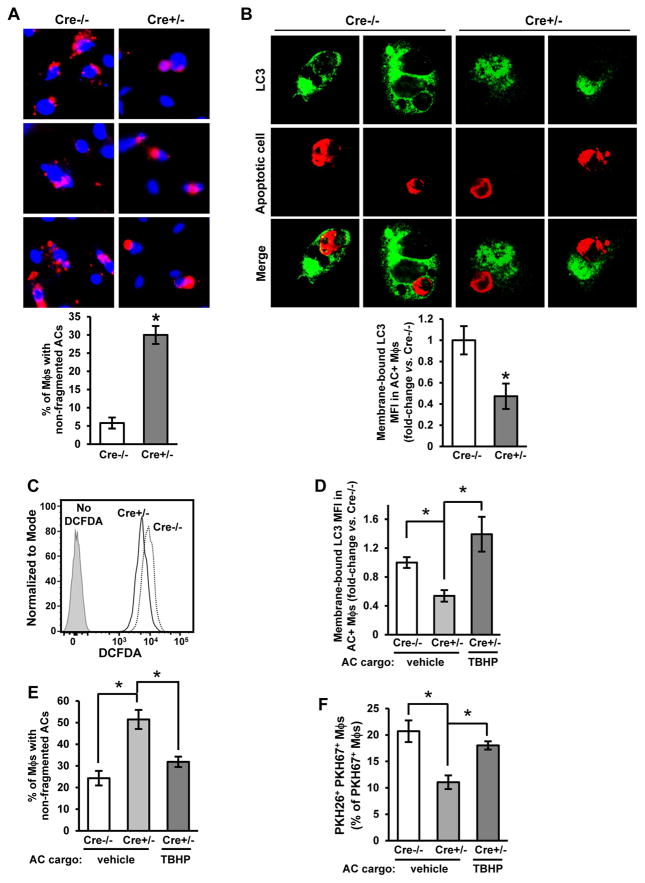
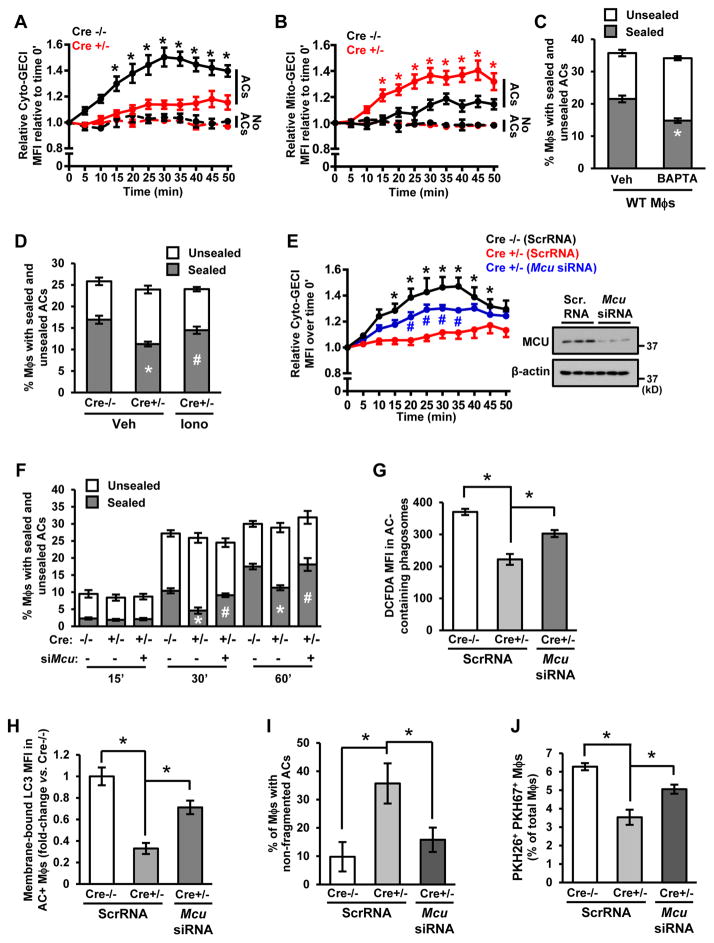
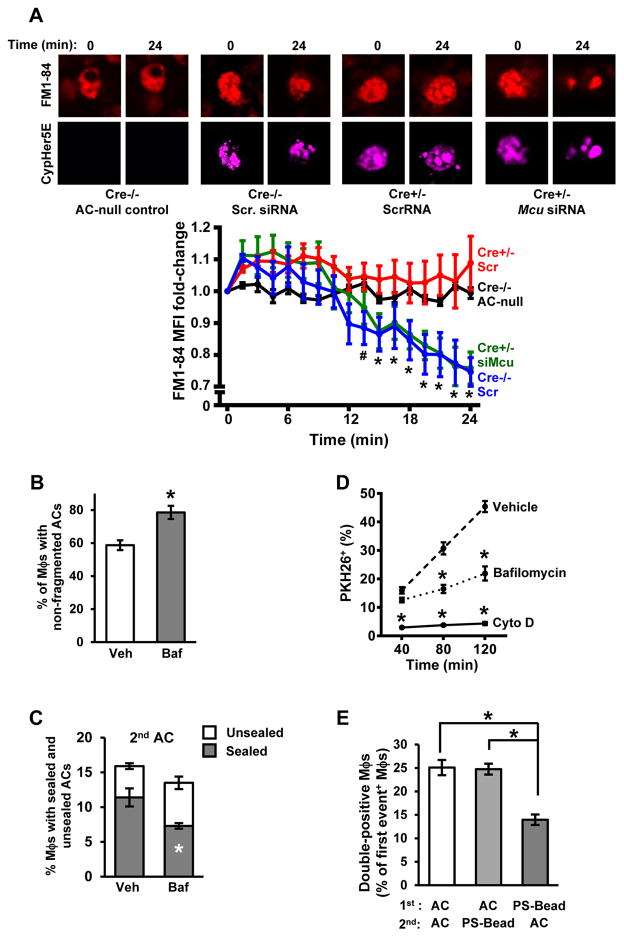
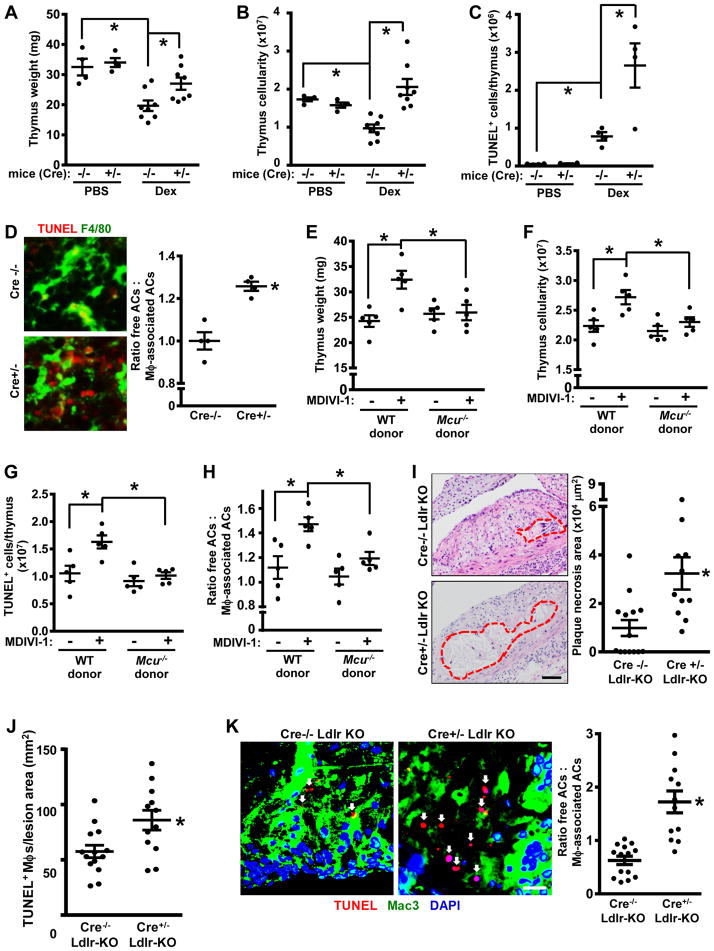
Clearance of apoptotic cells (ACs) by phagocytes (efferocytosis) prevents post-apoptotic necrosis and dampens inflammation. Defective efferocytosis drives important diseases, including atherosclerosis. For efficient efferocytosis, phagocytes must be able to internalize multiple ACs. We show here that uptake of multiple ACs by macrophages requires dynamin-related protein 1 (Drp1)-mediated mitochondrial fission, which is triggered by AC uptake. When mitochondrial fission is disabled, AC-induced increase in cytosolic calcium is blunted owing to mitochondrial calcium sequestration, and calcium-dependent phagosome formation around secondarily encountered ACs is impaired. These defects can be corrected by silencing the mitochondrial calcium uniporter (MCU). Mice lacking myeloid Drp1 showed defective efferocytosis and its pathologic consequences in the thymus after dexamethasone treatment and in advanced atherosclerotic lesions in fat-fed Ldlr-/- mice. Thus, mitochondrial fission in response to AC uptake is a critical process that enables macrophages to clear multiple ACs and to avoid the pathologic consequences of defective efferocytosis in vivo.
Keywords: DRP1; apoptotic cells; atherosclerosis; calcium signaling; efferocytosis; macrophage; mitochondrial dynamics; mitochondrial fission; phagocytosis.
Copyright © 2017 Elsevier Inc. All rights reserved.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
