

Prospects of Fine-Mapping Trait-Associated Genomic Regions by Using Summary Statistics from Genome-wide Association Studies
- PMID: 28942963
- PMCID: PMC5630179
- DOI: 10.1016/j.ajhg.2017.08.012
Prospects of Fine-Mapping Trait-Associated Genomic Regions by Using Summary Statistics from Genome-wide Association Studies
Abstract
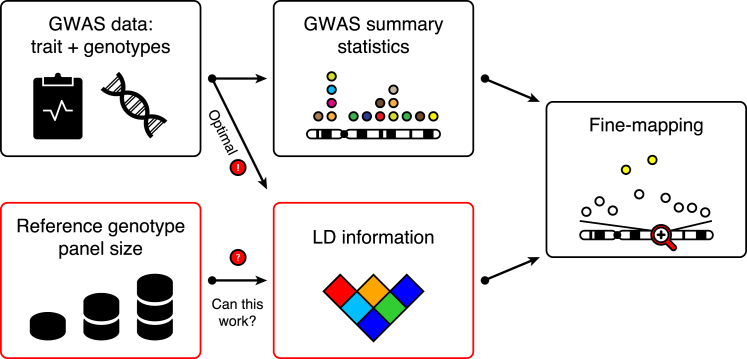
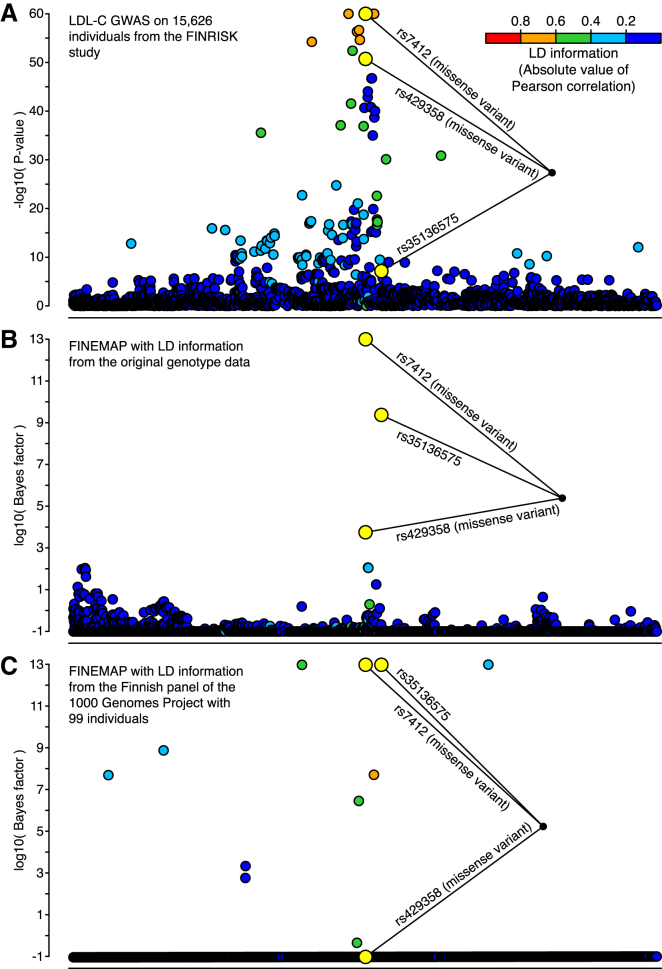
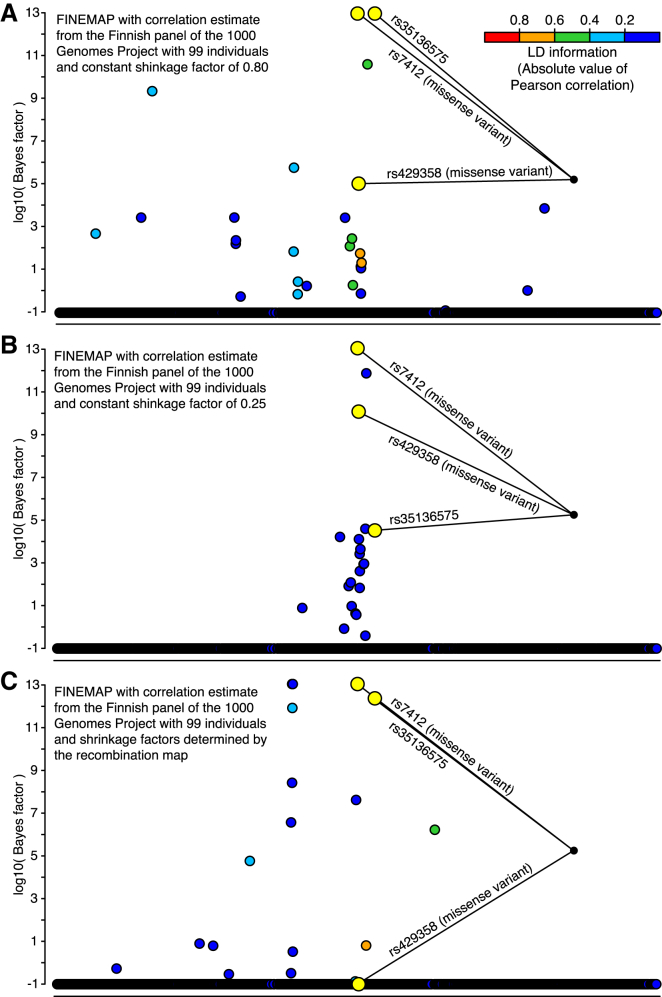
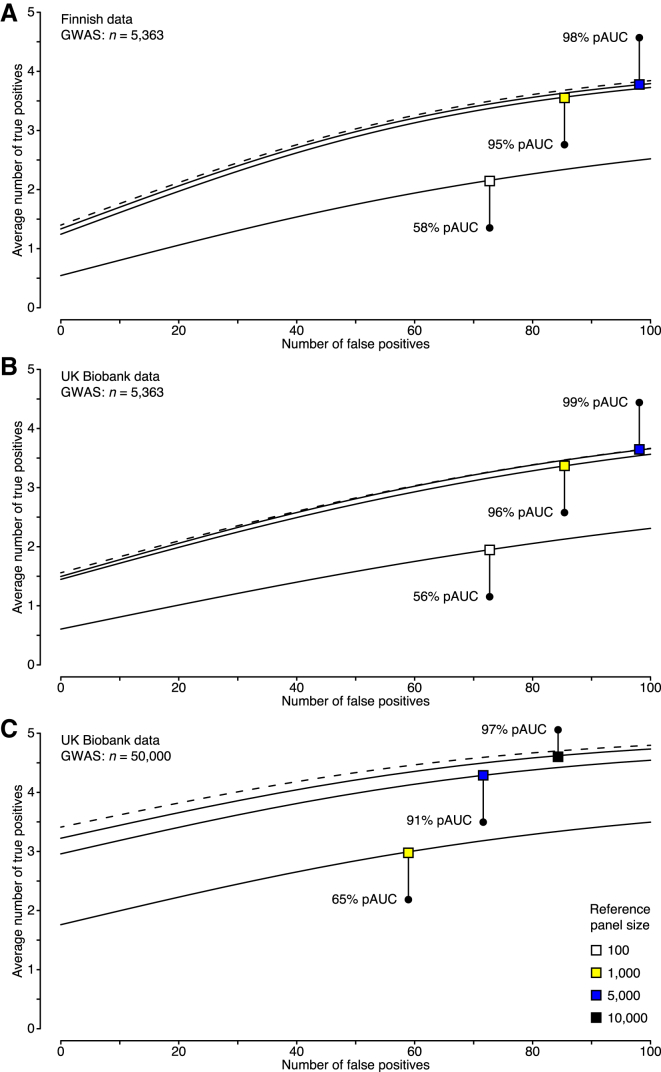
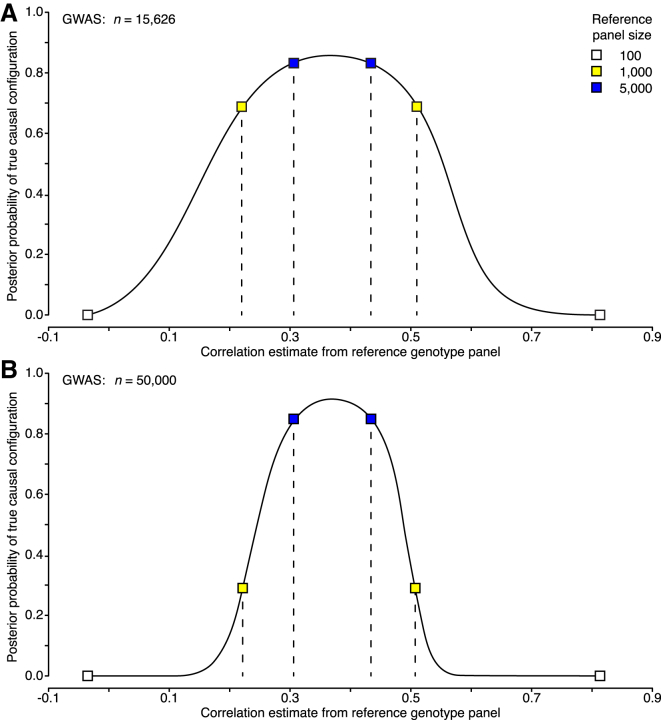
During the past few years, various novel statistical methods have been developed for fine-mapping with the use of summary statistics from genome-wide association studies (GWASs). Although these approaches require information about the linkage disequilibrium (LD) between variants, there has not been a comprehensive evaluation of how estimation of the LD structure from reference genotype panels performs in comparison with that from the original individual-level GWAS data. Using population genotype data from Finland and the UK Biobank, we show here that a reference panel of 1,000 individuals from the target population is adequate for a GWAS cohort of up to 10,000 individuals, whereas smaller panels, such as those from the 1000 Genomes Project, should be avoided. We also show, both theoretically and empirically, that the size of the reference panel needs to scale with the GWAS sample size; this has important consequences for the application of these methods in ongoing GWAS meta-analyses and large biobank studies. We conclude by providing software tools and by recommending practices for sharing LD information to more efficiently exploit summary statistics in genetics research.
Copyright © 2017 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Finucane H.K., Bulik-Sullivan B., Gusev A., Trynka G., Reshef Y., Loh P.R., Anttila V., Xu H., Zang C., Farh K., ReproGen Consortium. Schizophrenia Working Group of the Psychiatric Genomics Consortium. RACI Consortium Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 2015;47:1228–1235. - PMC - PubMed
-
- Bulik-Sullivan B., Finucane H.K., Anttila V., Gusev A., Day F.R., Loh P.R., Duncan L., Perry J.R., Patterson N., Robinson E.B., ReproGen Consortium. Psychiatric Genomics Consortium. Genetic Consortium for Anorexia Nervosa of the Wellcome Trust Case Control Consortium 3 An atlas of genetic correlations across human diseases and traits. Nat. Genet. 2015;47:1236–1241. - PMC - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
