

Next-generation sequencing reveals the mutational landscape of clinically diagnosed Usher syndrome: copy number variations, phenocopies, a predominant target for translational read-through, and PEX26 mutated in Heimler syndrome
- PMID: 28944237
- PMCID: PMC5606877
- DOI: 10.1002/mgg3.312
Next-generation sequencing reveals the mutational landscape of clinically diagnosed Usher syndrome: copy number variations, phenocopies, a predominant target for translational read-through, and PEX26 mutated in Heimler syndrome
Abstract
Background: Combined retinal degeneration and sensorineural hearing impairment is mostly due to autosomal recessive Usher syndrome (USH1: congenital deafness, early retinitis pigmentosa (RP); USH2: progressive hearing impairment, RP).
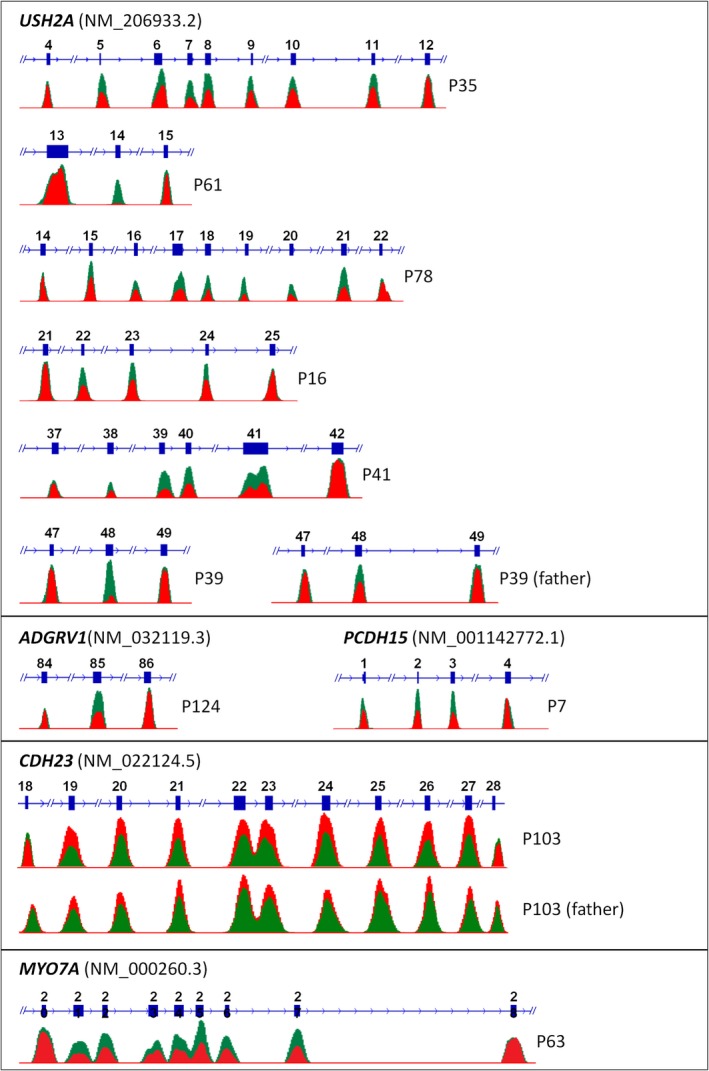
Methods: Sanger sequencing and NGS of 112 genes (Usher syndrome, nonsyndromic deafness, overlapping conditions), MLPA, and array-CGH were conducted in 138 patients clinically diagnosed with Usher syndrome.
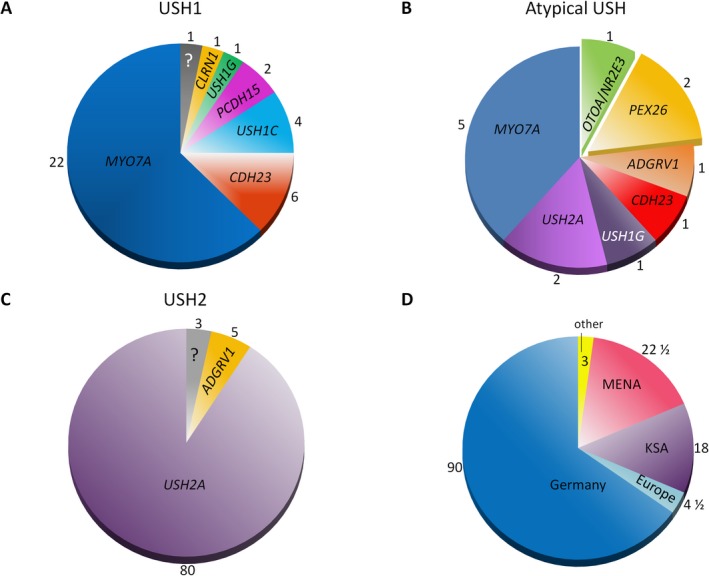
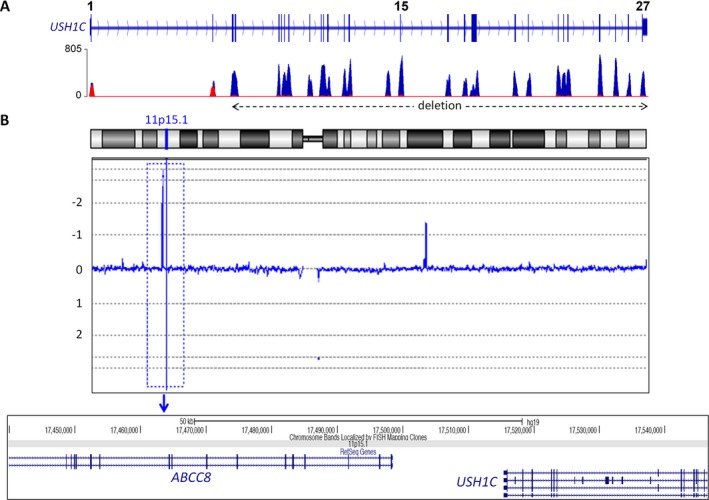
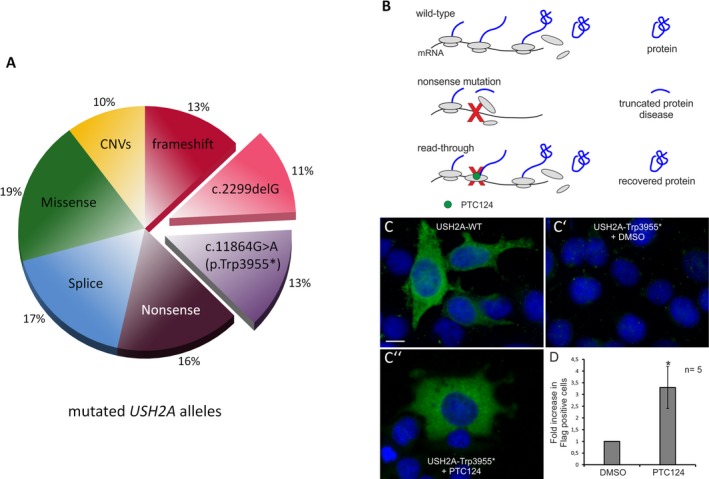
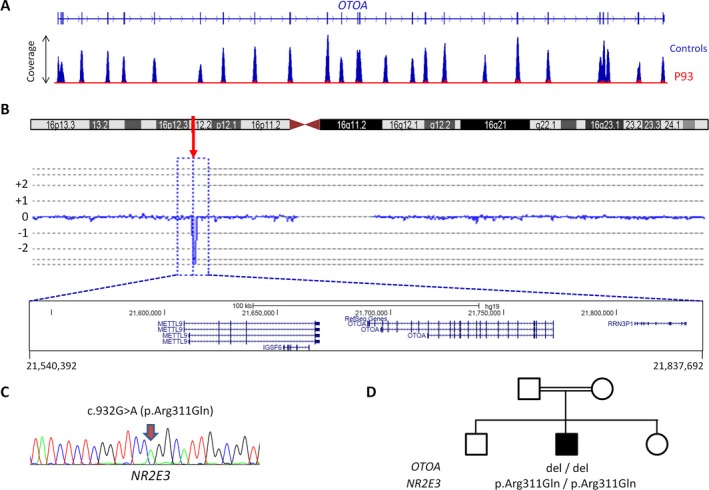
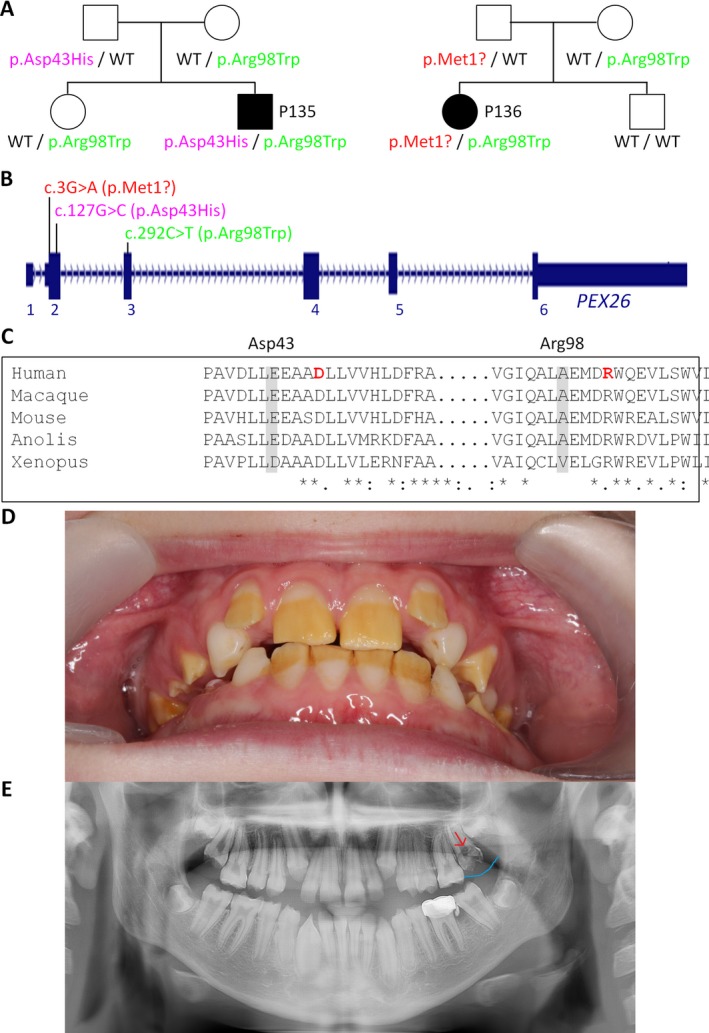
Results: A molecular diagnosis was achieved in 97% of both USH1 and USH2 patients, with biallelic mutations in 97% (USH1) and 90% (USH2), respectively. Quantitative readout reliably detected CNVs (confirmed by MLPA or array-CGH), qualifying targeted NGS as one tool for detecting point mutations and CNVs. CNVs accounted for 10% of identified USH2A alleles, often in trans to seemingly monoallelic point mutations. We demonstrate PTC124-induced read-through of the common p.Trp3955* nonsense mutation (13% of detected USH2A alleles), a potential therapy target. Usher gene mutations were found in most patients with atypical Usher syndrome, but the diagnosis was adjusted in case of double homozygosity for mutations in OTOA and NR2E3, genes implicated in isolated deafness and RP. Two patients with additional enamel dysplasia had biallelic PEX26 mutations, for the first time linking this gene to Heimler syndrome.
Conclusion: Targeted NGS not restricted to Usher genes proved beneficial in uncovering conditions mimicking Usher syndrome.
Keywords: Copy number variation; Heimler syndrome; Usher syndrome; next‐generation sequencing; phenocopies; translational read‐through.
Figures
References
-
- Adato, A. , Weston M. D., Berry A., Kimberling W. J., and Bonne‐Tamir A.. 2000. Three novel mutations and twelve polymorphisms identified in the USH2A gene in Israeli USH2 families. Hum. Mutat. 15:388. - PubMed
-
- Ahmed, Z. M. , Riazuddin S., Ahmad J., Bernstein S. 7., Guo Y., Sabar M. F., et al. 2003. PCDH15 is expressed in the neurosensory epithelium of the eye and ear and mutant alleles are responsible for both USH1F and DFNB23. Hum. Mol. Genet. 12:3215–3223. - PubMed
-
- Akoury, E. , El Zir E., Mansour A., Megarbane A., Majewski J., and Slim R.. 2011. A novel 5‐bp deletion in Clarin 1 in a family with Usher syndrome. Ophthalmic Genet. 32:245–249. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
