

Benchmarking viromics: an in silico evaluation of metagenome-enabled estimates of viral community composition and diversity
- PMID: 28948103
- PMCID: PMC5610896
- DOI: 10.7717/peerj.3817
Benchmarking viromics: an in silico evaluation of metagenome-enabled estimates of viral community composition and diversity
Abstract
Background: Viral metagenomics (viromics) is increasingly used to obtain uncultivated viral genomes, evaluate community diversity, and assess ecological hypotheses. While viromic experimental methods are relatively mature and widely accepted by the research community, robust bioinformatics standards remain to be established. Here we used in silico mock viral communities to evaluate the viromic sequence-to-ecological-inference pipeline, including (i) read pre-processing and metagenome assembly, (ii) thresholds applied to estimate viral relative abundances based on read mapping to assembled contigs, and (iii) normalization methods applied to the matrix of viral relative abundances for alpha and beta diversity estimates.
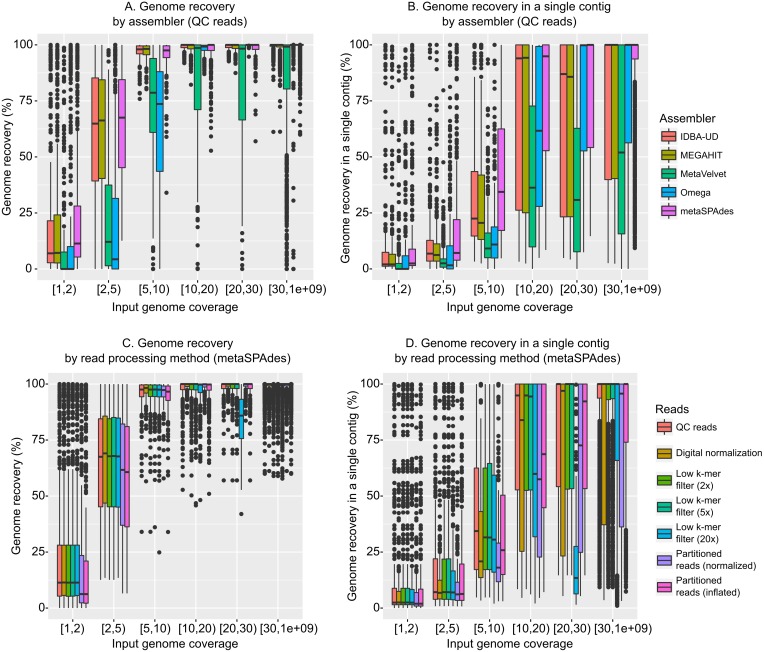
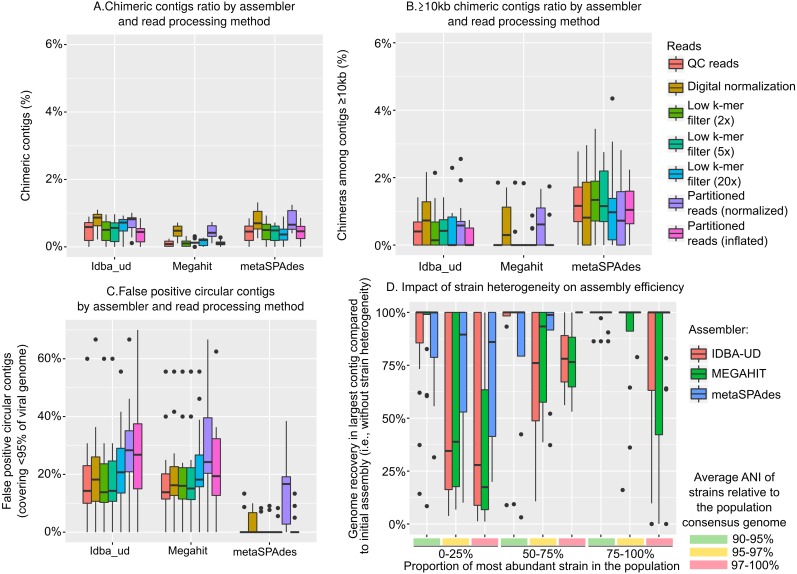
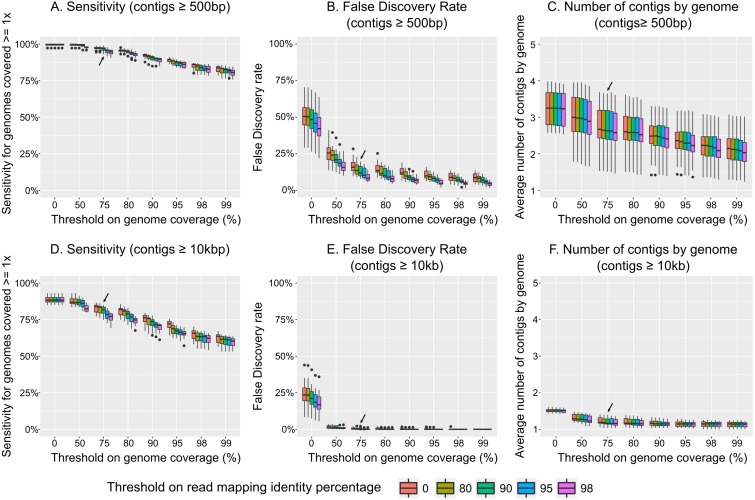
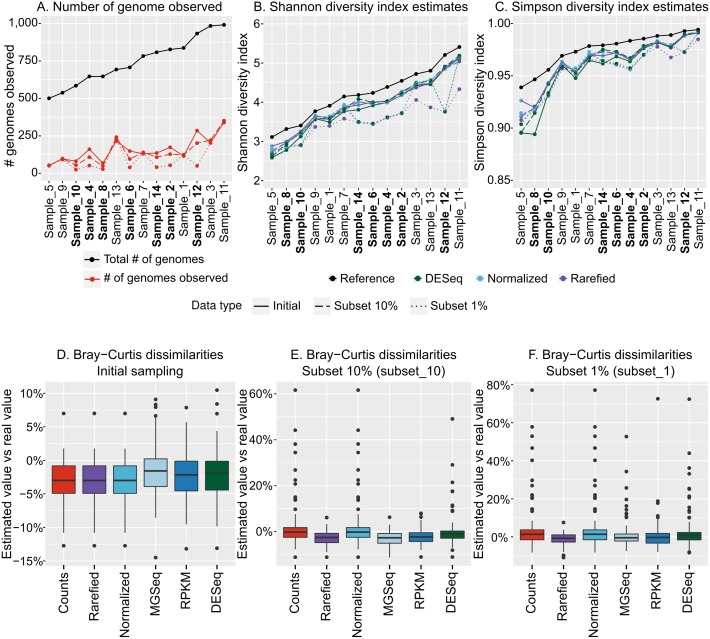
Results: Tools specifically designed for metagenomes, specifically metaSPAdes, MEGAHIT, and IDBA-UD, were the most effective at assembling viromes. Read pre-processing, such as partitioning, had virtually no impact on assembly output, but may be useful when hardware is limited. Viral populations with 2-5 × coverage typically assembled well, whereas lesser coverage led to fragmented assembly. Strain heterogeneity within populations hampered assembly, especially when strains were closely related (average nucleotide identity, or ANI ≥97%) and when the most abundant strain represented <50% of the population. Viral community composition assessments based on read recruitment were generally accurate when the following thresholds for detection were applied: (i) ≥10 kb contig lengths to define populations, (ii) coverage defined from reads mapping at ≥90% identity, and (iii) ≥75% of contig length with ≥1 × coverage. Finally, although data are limited to the most abundant viruses in a community, alpha and beta diversity patterns were robustly estimated (±10%) when comparing samples of similar sequencing depth, but more divergent (up to 80%) when sequencing depth was uneven across the dataset. In the latter cases, the use of normalization methods specifically developed for metagenomes provided the best estimates.
Conclusions: These simulations provide benchmarks for selecting analysis cut-offs and establish that an optimized sample-to-ecological-inference viromics pipeline is robust for making ecological inferences from natural viral communities. Continued development to better accessing RNA, rare, and/or diverse viral populations and improved reference viral genome availability will alleviate many of viromics remaining limitations.
Keywords: Assembly; Benchmarks; Metagenome; Viral ecology; Virome; Virus.
Conflict of interest statement
The authors declare there are no competing interests.
Figures
References
-
- Allers E, Moraru C, Duhaime MB, Beneze E, Solonenko N, Canosa JB, Amann R, Sullivan MB. Single-cell and population level viral infection dynamics revealed by phageFISH, a method to visualize intracellular and free viruses. Environmental Microbiology. 2013;15:2306–2318. doi: 10.1111/1462-2920.12100. - DOI - PMC - PubMed
-
- Angly FE, Willner D, Prieto-Davó A, Edwards RA, Schmieder R, Vega-Thurber R, Antonopoulos DA, Barott K, Cottrell MT, Desnues C, Dinsdale EA, Furlan M, Haynes M, Henn MR, Hu Y, Kirchman DL, McDole T, McPherson JD, Meyer F, Miller RM, Mundt E, Naviaux RK, Rodriguez-Mueller B, Stevens R, Wegley L, Zhang L, Zhu B, Rohwer F. The GAAS metagenomic tool and its estimations of viral and microbial average genome size in four major biomes. PLOS Computational Biology. 2009;5:e1000593. doi: 10.1371/journal.pcbi.1000593. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
