

Sexually divergent DNA methylation patterns with hippocampal aging
- PMID: 28948711
- PMCID: PMC5676057
- DOI: 10.1111/acel.12681
Sexually divergent DNA methylation patterns with hippocampal aging
Abstract
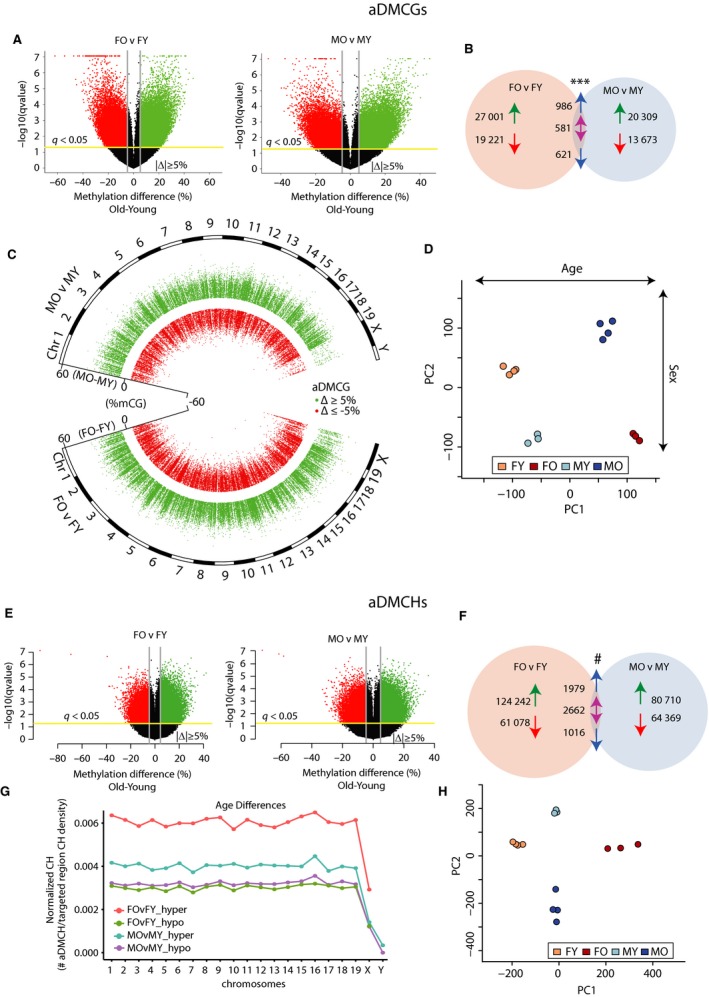
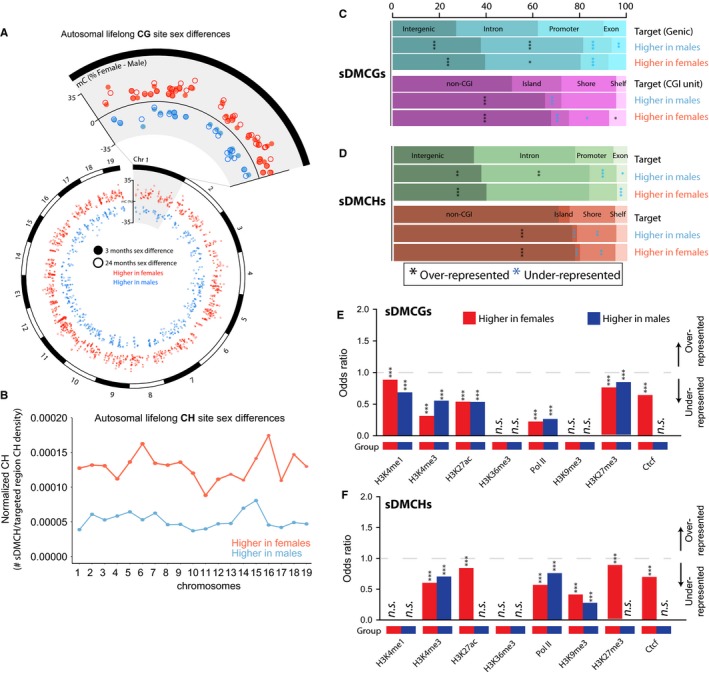
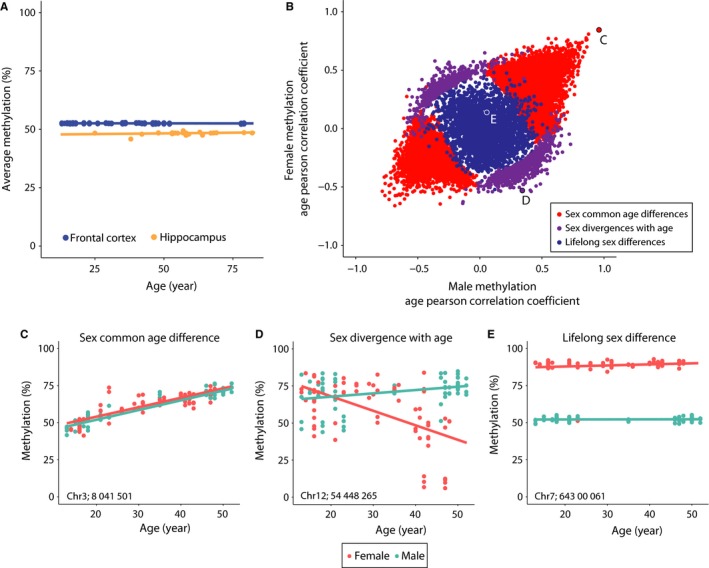
DNA methylation is a central regulator of genome function, and altered methylation patterns are indicative of biological aging and mortality. Age-related cellular, biochemical, and molecular changes in the hippocampus lead to cognitive impairments and greater vulnerability to neurodegenerative disease that varies between the sexes. The role of hippocampal epigenomic changes with aging in these processes is unknown as no genome-wide analyses of age-related methylation changes have considered the factor of sex in a controlled animal model. High-depth, genome-wide bisulfite sequencing of young (3 month) and old (24 month) male and female mouse hippocampus revealed that while total genomic methylation amounts did not change with aging, specific sites in CG and non-CG (CH) contexts demonstrated age-related increases or decreases in methylation that were predominantly sexually divergent. Differential methylation with age for both CG and CH sites was enriched in intergenic and intronic regions and under-represented in promoters, CG islands, and specific enhancer regions in both sexes, suggesting that certain genomic elements are especially labile with aging, even if the exact genomic loci altered are predominantly sex-specific. Lifelong sex differences in autosomal methylation at CG and CH sites were also observed. The lack of genome-wide hypomethylation, sexually divergent aging response, and autosomal sex differences at CG sites was confirmed in human data. These data reveal sex as a previously unappreciated central factor of hippocampal epigenomic changes with aging. In total, these data demonstrate an intricate regulation of DNA methylation with aging by sex, cytosine context, genomic location, and methylation level.
Keywords: DNA methylation; aging; divergence; epigenetics; hippocampus; sex differences.
© 2017 The Authors. Aging Cell published by the Anatomical Society and John Wiley & Sons Ltd.
Figures


References
-
- Ashapkin VV, Kutueva LI, Vanyushin BF (2015) Aging epigenetics: accumulation of errors or realization of a specific program? Biochemistry (Mosc) 80, 1406–1417. - PubMed
-
- Chow HM, Herrup K (2015) Genomic integrity and the ageing brain. Nat. Rev. Neurosci. 16, 672–684. - PubMed
-
- Cole JJ, Robertson NA, Rather MI, Thomson JP, McBryan T, Sproul D, Wang T, Brock C, Clark W, Ideker T, Meehan RR, Miller RA, Brown‐Borg HM, Adams PD (2017) Diverse interventions that extend mouse lifespan suppress shared age‐associated epigenetic changes at critical gene regulatory regions. Genome Biol. 18, 58. - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
