

Iterative Molecular Dynamics-Rosetta Membrane Protein Structure Refinement Guided by Cryo-EM Densities
- PMID: 28949136
- PMCID: PMC5642286
- DOI: 10.1021/acs.jctc.7b00464
Iterative Molecular Dynamics-Rosetta Membrane Protein Structure Refinement Guided by Cryo-EM Densities
Abstract
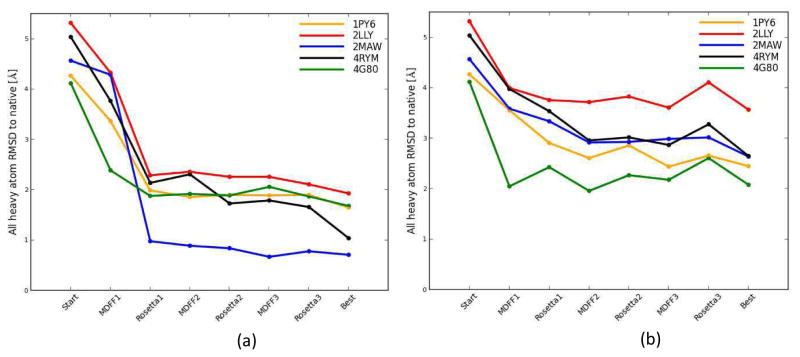
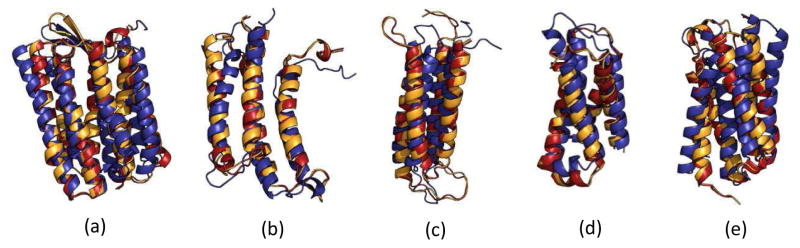
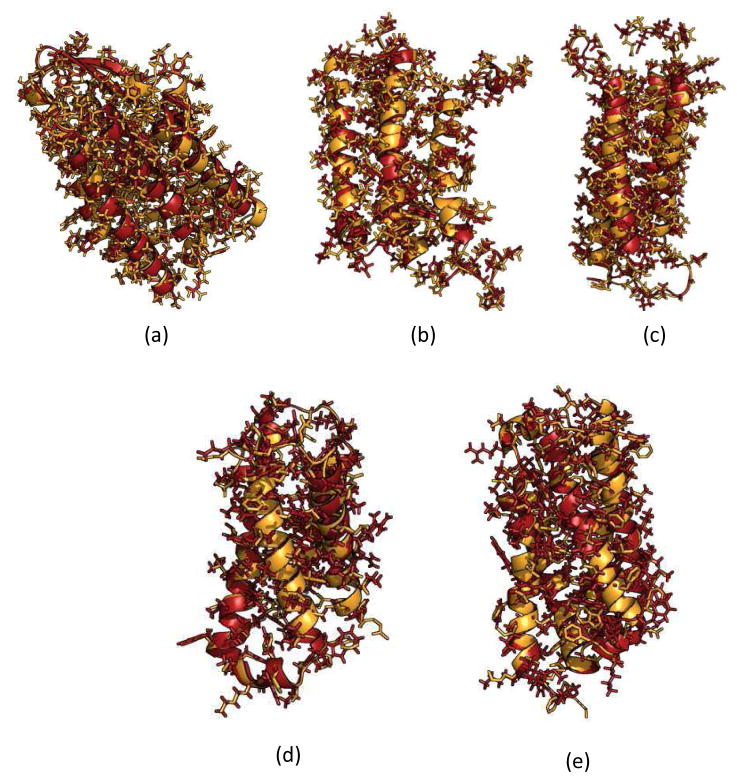
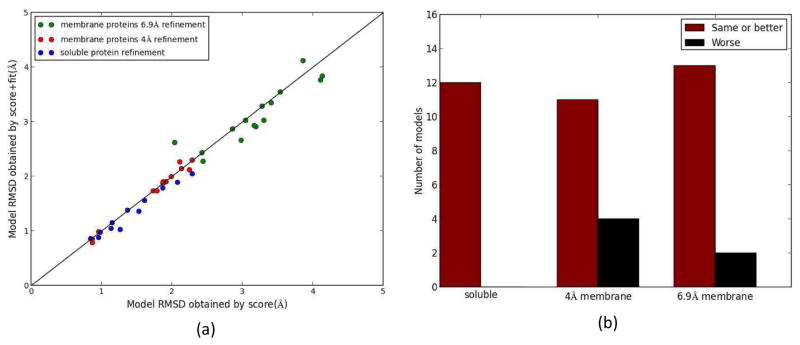
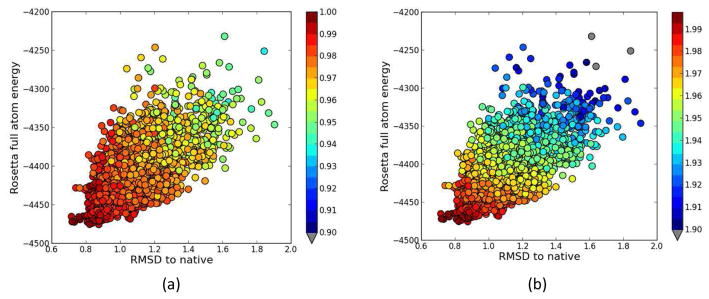
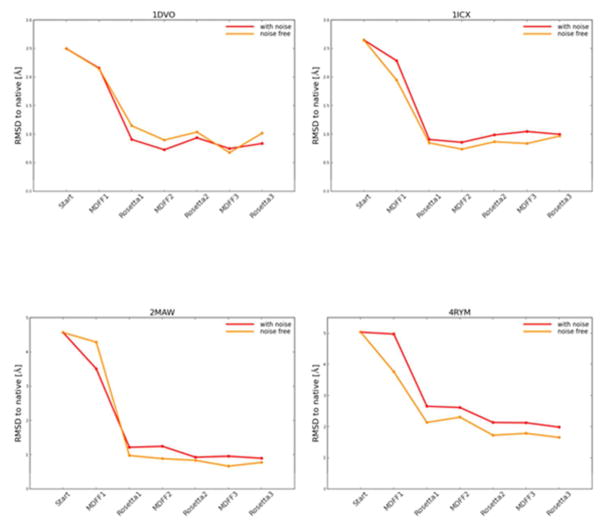
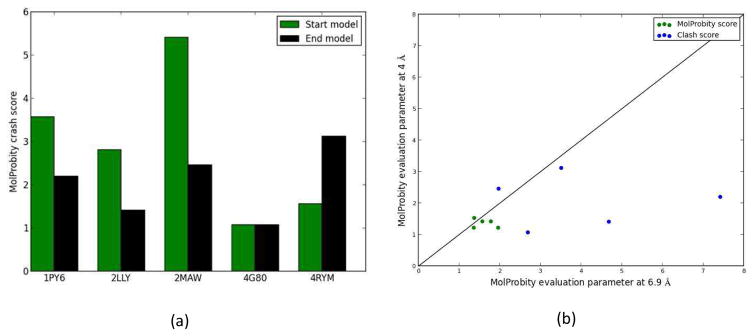
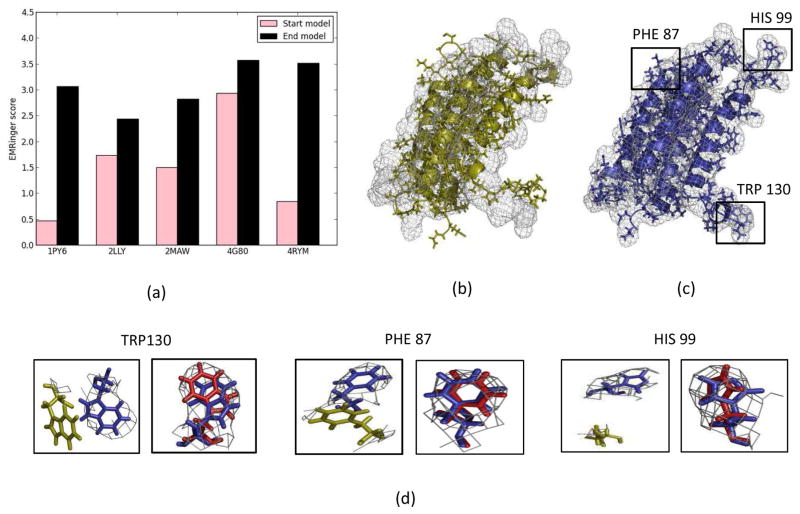
Knowing atomistic details of proteins is essential not only for the understanding of protein function but also for the development of drugs. Experimental methods such as X-ray crystallography, NMR, and cryo-electron microscopy (cryo-EM) are the preferred forms of protein structure determination and have achieved great success over the most recent decades. Computational methods may be an alternative when experimental techniques fail. However, computational methods are severely limited when it comes to predicting larger macromolecule structures with little sequence similarity to known structures. The incorporation of experimental restraints in computational methods is becoming increasingly important to more reliably predict protein structure. One such experimental input used in structure prediction and refinement is cryo-EM densities. Recent advances in cryo-EM have arguably revolutionized the field of structural biology. Our previously developed cryo-EM-guided Rosetta-MD protocol has shown great promise in the refinement of soluble protein structures. In this study, we extended cryo-EM density-guided iterative Rosetta-MD to membrane proteins. We also improved the methodology in general by picking models based on a combination of their score and fit-to-density during the Rosetta model selection. By doing so, we have been able to pick models superior to those with the previous selection based on Rosetta score only and we have been able to further improve our previously refined models of soluble proteins. The method was tested with five membrane spanning protein structures. By applying density-guided Rosetta-MD iteratively we were able to refine the predicted structures of these membrane proteins to atomic resolutions. We also showed that the resolution of the density maps determines the improvement and quality of the refined models. By incorporating high-resolution density maps (∼4 Å), we were able to more significantly improve the quality of the models than when medium-resolution maps (6.9 Å) were used. Beginning from an average starting structure root mean square deviation (RMSD) to native of 4.66 Å, our protocol was able to refine the structures to bring the average refined structure RMSD to 1.66 Å when 4 Å density maps were used. The protocol also successfully refined the HIV-1 CTD guided by an experimental 5 Å density map.
Figures
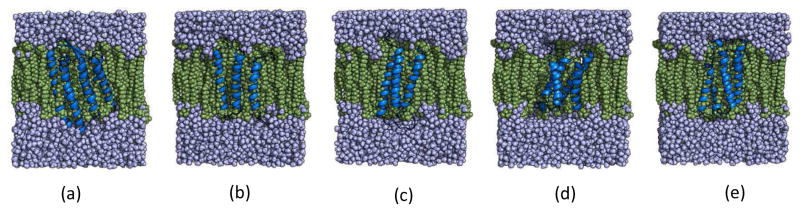
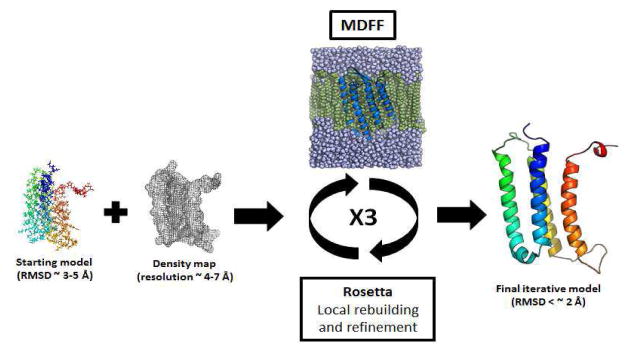
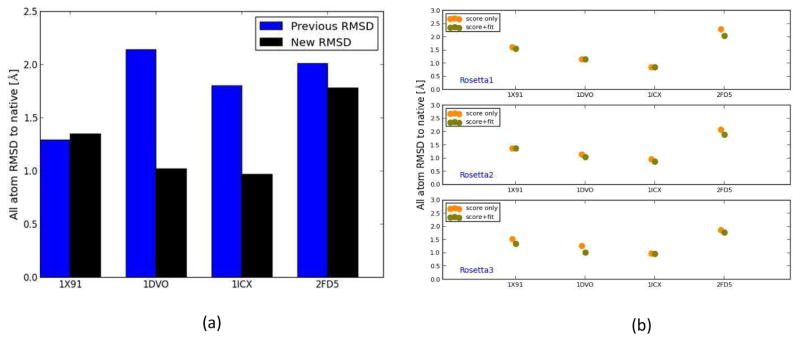
References
-
- Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P. The shape and structure of proteins. 2002
-
- Jacobson M, Sali A. Comparative protein structure modeling and its applications to drug discovery. Annual reports in medicinal chemistry. 2004;39:259–276.
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources