

PKCζ as a promising therapeutic target for TNFα-induced inflammatory disorders in chronic cutaneous wounds
- PMID: 28949382
- PMCID: PMC5627866
- DOI: 10.3892/ijmm.2017.3144
PKCζ as a promising therapeutic target for TNFα-induced inflammatory disorders in chronic cutaneous wounds
Abstract
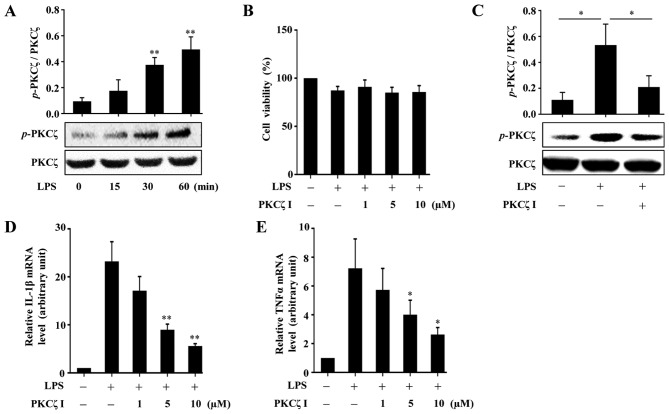
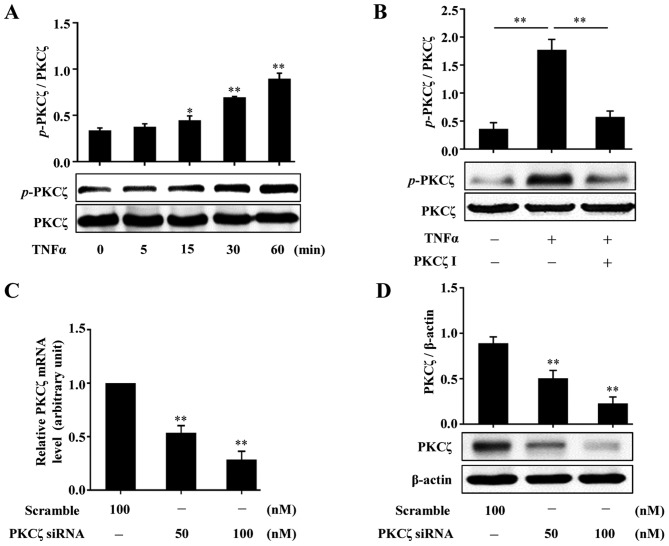
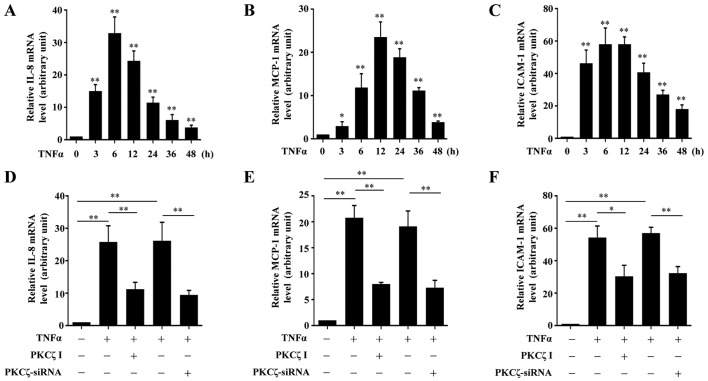
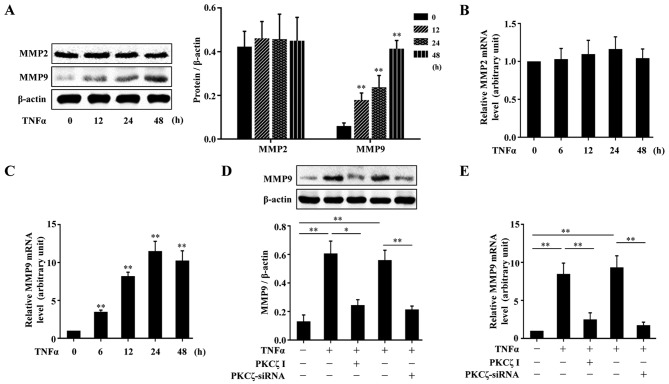
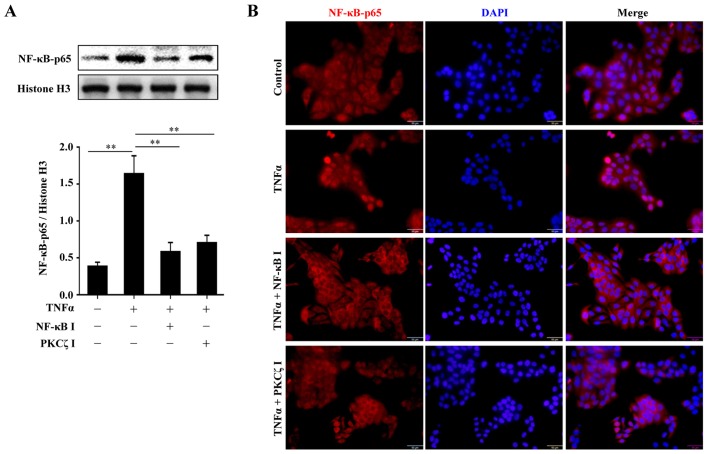
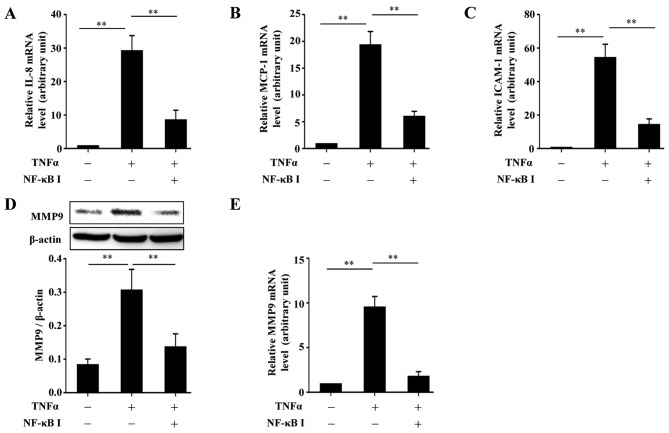
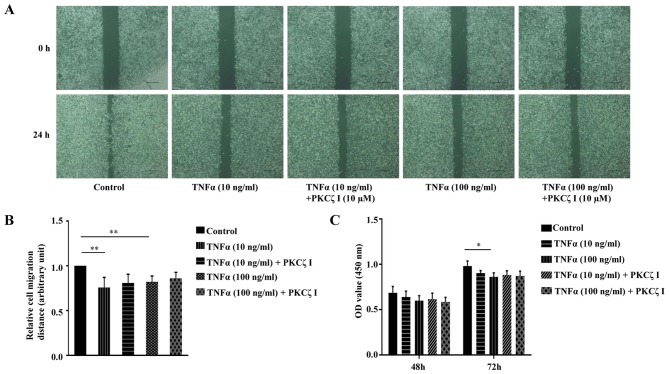
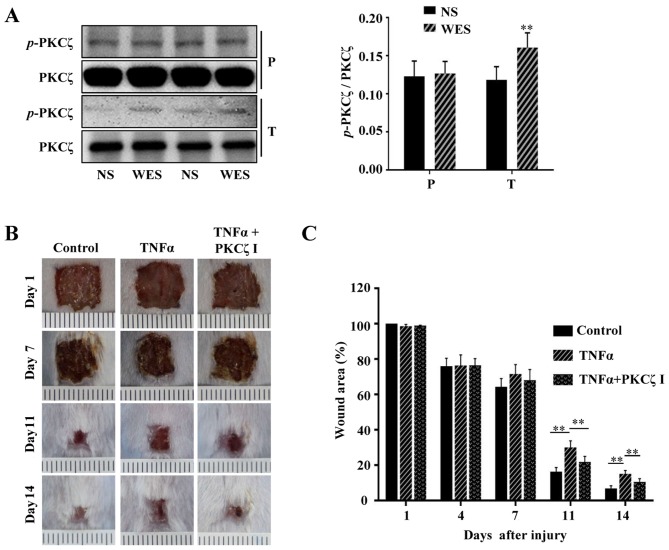
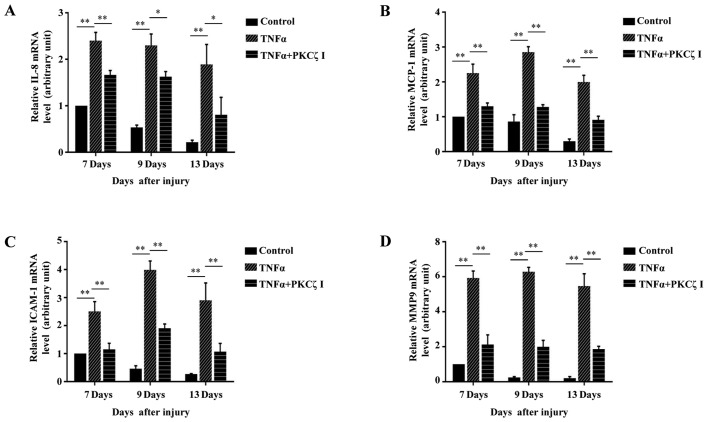
Protein kinase Cζ (PKCζ) is a member of the atypical protein kinase C family. Its roles in macrophages or skin-resident keratinocytes have not been fully evaluated. In this study, we provide evidence that PKCζ mediates lipopolysaccharide (LPS)-induced tumor necrosis factor α (TNFα) gene expression in the mouse macrophage cell line, RAW264.7. TNFα has been proven to be one of the main culprits of chronic wounds and impaired acute wounds, which are characterized by excessive inflammation, enhanced proteolysis and reduced matrix deposition. Among the multiple effects of TNFα on keratinocytes, the induction of chemokines which are indispensable factors involved in the massive infiltration of various inflammatory cells into skin lesions serves as a crucial mechanism. In the present study, we found that PKCζ inhibitor or its specific siRNA inhibited the TNFα-induced upregulation in the levels of the chemokines, interleukin (IL)-8, monocyte chemotactic protein-1 (MCP-1) and intercellular cell adhesion molecule-1 (ICAM-1) in HaCaT keratinocytes. Moreover, under a disrupted inflammatory environment, activated keratinocytes can synthesize large amounts of matrix metalloproteinases (MMP), which has a negative effect on tissue remodeling. We discovered that TNFα promoted the expression of MMP9 in a PKCζ-dependent manner. Further experiments revealed that nuclear factor-κB (NF-κB) was a key downstream molecule of PKCζ. In addition, as shown in vitro, PKCζ was not involved in the TNFα-induced decrease in HaCaT cell migration and proliferation. In vivo experiments demonstrated that TNFα-induced wound closure impairment and inflammatory disorders were significantly attenuated in the PKCζ inhibitor group. On the whole, our findings suggest that PKCζ is a crucial regulator in LPS- or TNFα-induced inflammatory responses in RAW264.7 cells and HaCaT keratinocytes, and that PKCζ/NF-κB signaling may be a potential target for interventional therapy for TNFα-induced skin inflammatory injury.
Figures
Similar articles
-
Protein kinase Czeta (PKCzeta) regulates ocular inflammation and apoptosis in endotoxin-induced uveitis (EIU): signaling molecules involved in EIU resolution by PKCzeta inhibitor and interleukin-13.Am J Pathol. 2007 Apr;170(4):1241-57. doi: 10.2353/ajpath.2007.060236. Am J Pathol. 2007. PMID: 17392164 Free PMC article.
-
Upregulation of MMP-9 production by TNFalpha in keratinocytes and its attenuation by vitamin D.J Cell Physiol. 2010 Mar;222(3):729-37. doi: 10.1002/jcp.22004. J Cell Physiol. 2010. PMID: 20020446
-
Suppression of LPS-induced NF-κB activity in macrophages by the synthetic aurone, (Z)-2-((5-(hydroxymethyl) furan-2-yl) methylene) benzofuran-3(2H)-one.Int Immunopharmacol. 2017 Feb;43:116-128. doi: 10.1016/j.intimp.2016.12.004. Epub 2016 Dec 16. Int Immunopharmacol. 2017. PMID: 27988459
-
Tumor Necrosis Factor-Alpha: Ally and Enemy in Protean Cutaneous Sceneries.Int J Mol Sci. 2024 Jul 16;25(14):7762. doi: 10.3390/ijms25147762. Int J Mol Sci. 2024. PMID: 39063004 Free PMC article. Review.
-
Mechanosignaling in bone health, trauma and inflammation.Antioxid Redox Signal. 2014 Feb 20;20(6):970-85. doi: 10.1089/ars.2013.5467. Epub 2013 Aug 12. Antioxid Redox Signal. 2014. PMID: 23815527 Free PMC article. Review.
Cited by
-
Artemisia anomala Herba Alleviates 2,4-Dinitrochlorobenzene-Induced Atopic Dermatitis-Like Skin Lesions in Mice and the Production of Pro-Inflammatory Mediators in Tumor Necrosis Factor Alpha-/Interferon Gamma-Induced HaCaT Cells.Molecules. 2021 Sep 6;26(17):5427. doi: 10.3390/molecules26175427. Molecules. 2021. PMID: 34500860 Free PMC article.
-
Recent advances in anti-inflammatory active components and action mechanisms of natural medicines.Inflammopharmacology. 2023 Dec;31(6):2901-2937. doi: 10.1007/s10787-023-01369-9. Epub 2023 Nov 10. Inflammopharmacology. 2023. PMID: 37947913 Review.
-
Epidermal Stem Cells in Hair Follicle Cycling and Skin Regeneration: A View From the Perspective of Inflammation.Front Cell Dev Biol. 2020 Nov 9;8:581697. doi: 10.3389/fcell.2020.581697. eCollection 2020. Front Cell Dev Biol. 2020. PMID: 33240882 Free PMC article. Review.
-
Macrophages as a therapeutic target to promote diabetic wound healing.Mol Ther. 2022 Sep 7;30(9):2891-2908. doi: 10.1016/j.ymthe.2022.07.016. Epub 2022 Aug 2. Mol Ther. 2022. PMID: 35918892 Free PMC article. Review.
-
Palmitoylation of PKCδ by ZDHHC5 in hypothalamic microglia presents as a therapeutic target for fatty liver disease.Theranostics. 2024 Jan 1;14(3):988-1009. doi: 10.7150/thno.89602. eCollection 2024. Theranostics. 2024. PMID: 38250049 Free PMC article.
References
-
- Wetzler C, Kämpfer H, Stallmeyer B, Pfeilschifter J, Frank S. Large and sustained induction of chemokines during impaired wound healing in the genetically diabetic mouse: Prolonged persistence of neutrophils and macrophages during the late phase of repair. J Invest Dermatol. 2000;115:245–253. doi: 10.1046/j.1523-1747.2000.00029.x. - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
