

Mechanisms of Acquired Resistance to BRAF V600E Inhibition in Colon Cancers Converge on RAF Dimerization and Are Sensitive to Its Inhibition
- PMID: 28951457
- PMCID: PMC5712250
- DOI: 10.1158/0008-5472.CAN-17-0768
Mechanisms of Acquired Resistance to BRAF V600E Inhibition in Colon Cancers Converge on RAF Dimerization and Are Sensitive to Its Inhibition
Abstract
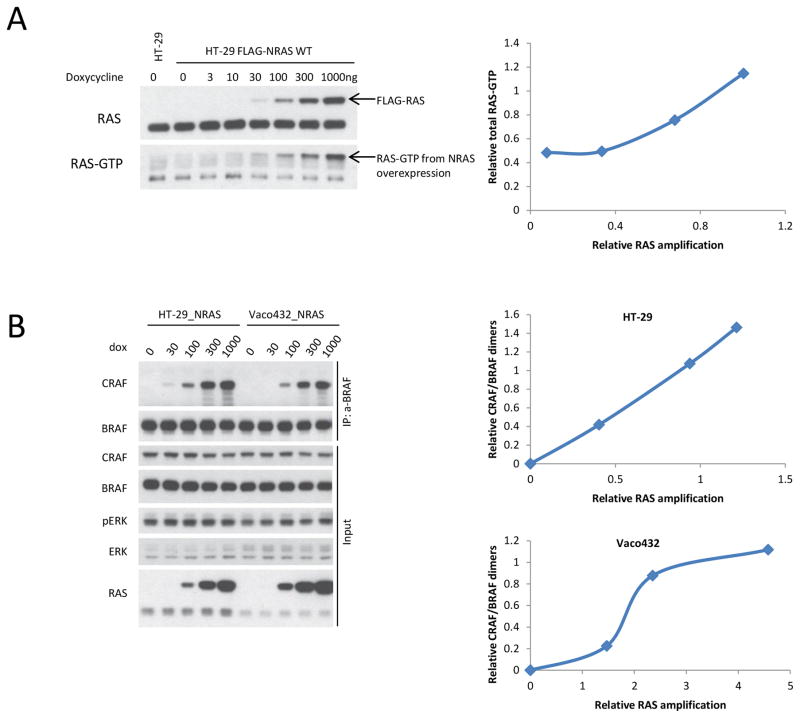
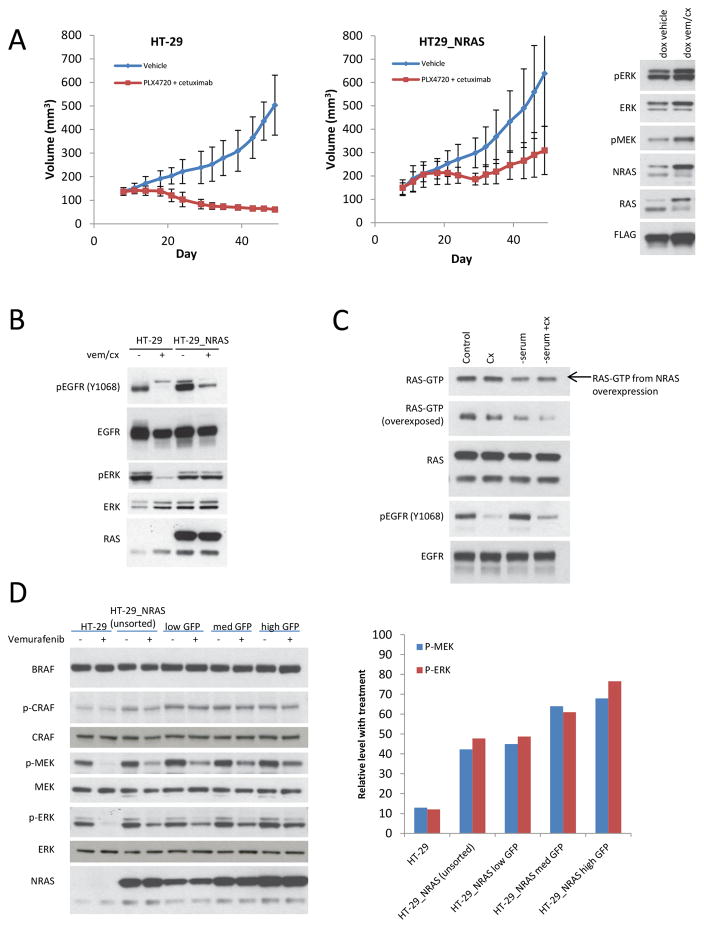
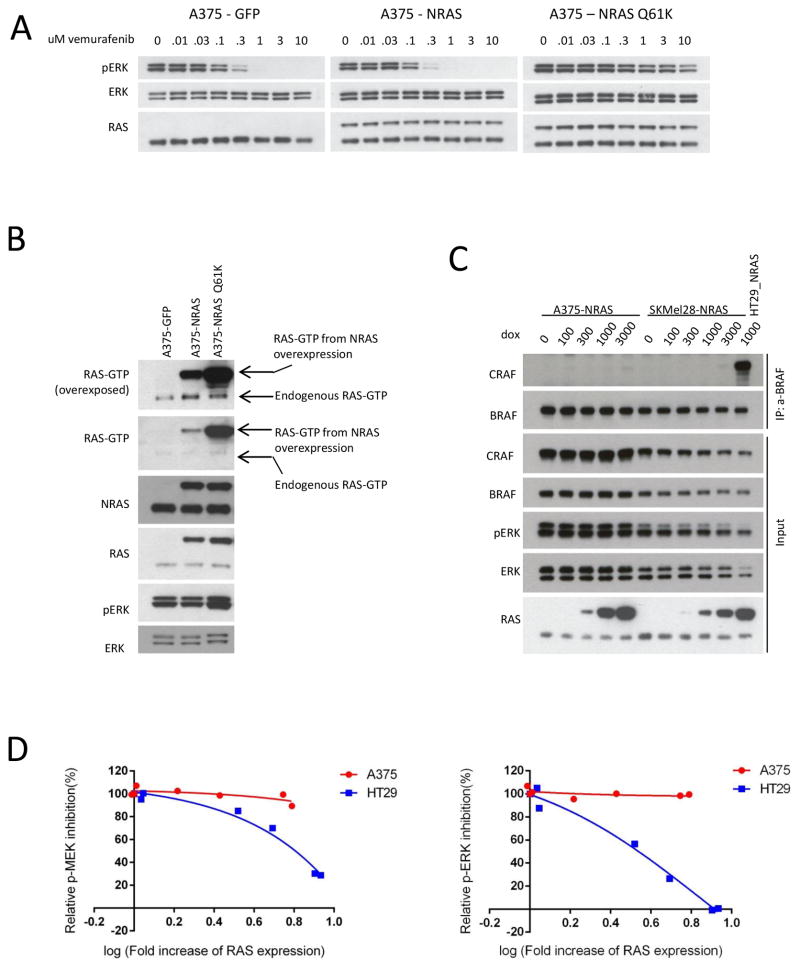
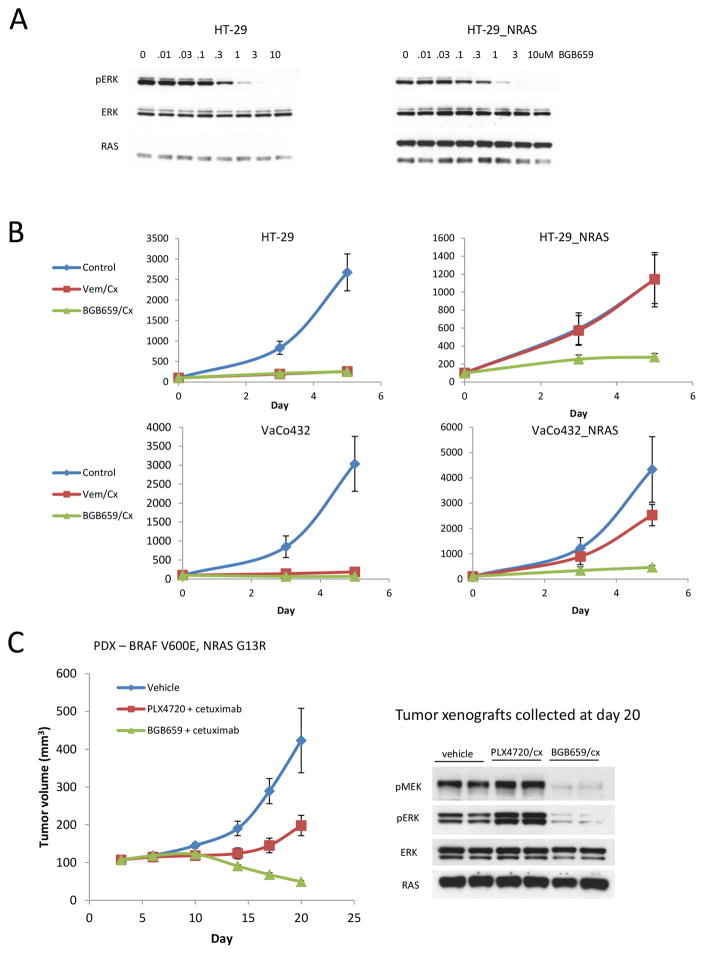
BRAF V600E colorectal cancers are insensitive to RAF inhibitor monotherapy due to feedback reactivation of receptor tyrosine kinase signaling. Combined RAF and EGFR inhibition exerts a therapeutic effect, but resistance invariably develops through undefined mechanisms. In this study, we determined that colorectal cancer progression specimens invariably harbored lesions in elements of the RAS-RAF-MEK-ERK pathway. Genetic amplification of wild-type RAS was a recurrent mechanism of resistance in colorectal cancer patients that was not seen in similarly resistant melanomas. We show that wild-type RAS amplification increases receptor tyrosine kinase-dependent activation of RAS more potently in colorectal cancer than in melanoma and causes resistance only in the former. Currently approved RAF inhibitors inhibit RAF monomers but not dimers. All the drug-resistant lesions we identified activate BRAF V600E dimerization directly or by elevating RAS-GTP. Overall, our results show that mechanisms of resistance converge on formation of RAF dimers and that inhibiting EGFR and RAF dimers can effectively suppress ERK-driven growth of resistant colorectal cancer. Cancer Res; 77(23); 6513-23. ©2017 AACR.
©2017 American Association for Cancer Research.
Figures
References
-
- Weber CK, Slupsky JR, Kalmes HA, Rapp UR. Active Ras induces heterodimerization of cRaf and BRaf. Cancer Res. 2001;61:3595–8. - PubMed
-
- Dougherty MK, Muller J, Ritt DA, Zhou M, Zhou XZ, Copeland TD, et al. Regulation of Raf-1 by direct feedback phosphorylation. Mol Cell. 2005;17:215–24. - PubMed
-
- Douville E, Downward J. EGF induced SOS phosphorylation in PC12 cells involves P90 RSK-2. Oncogene. 1997;15:373–83. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
