

Systematic Discovery of Archaeal Transcription Factor Functions in Regulatory Networks through Quantitative Phenotyping Analysis
- PMID: 28951888
- PMCID: PMC5605881
- DOI: 10.1128/mSystems.00032-17
Systematic Discovery of Archaeal Transcription Factor Functions in Regulatory Networks through Quantitative Phenotyping Analysis
Abstract
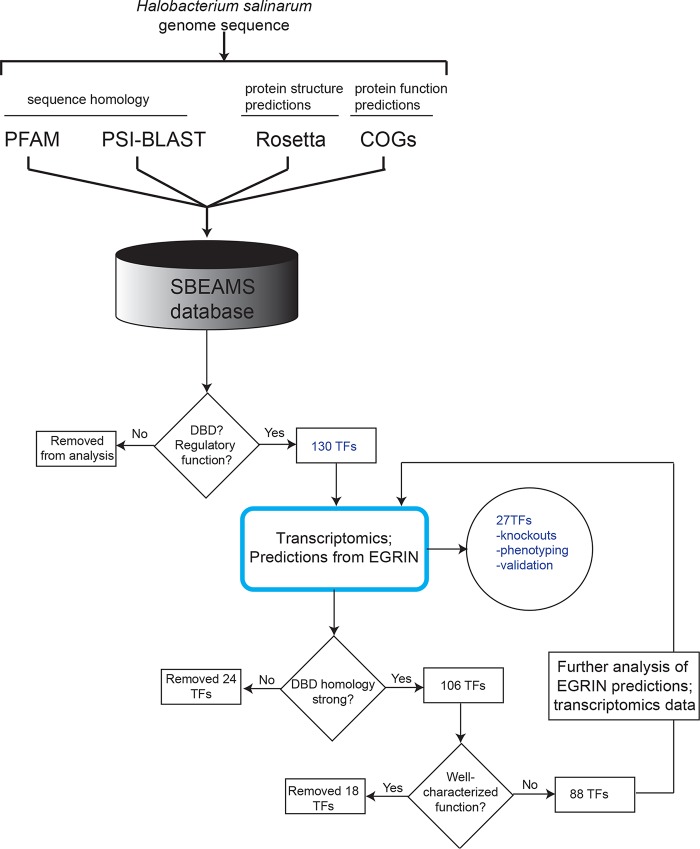
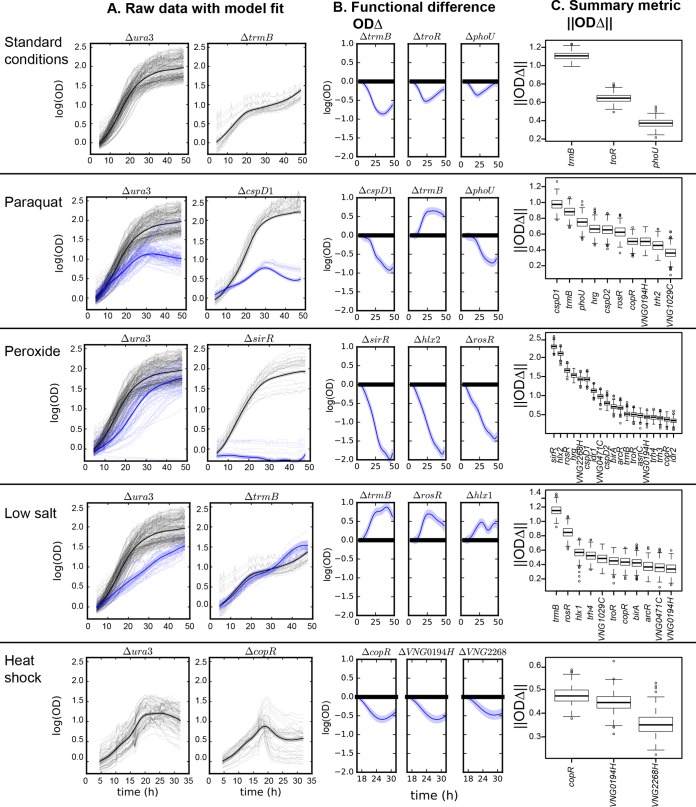
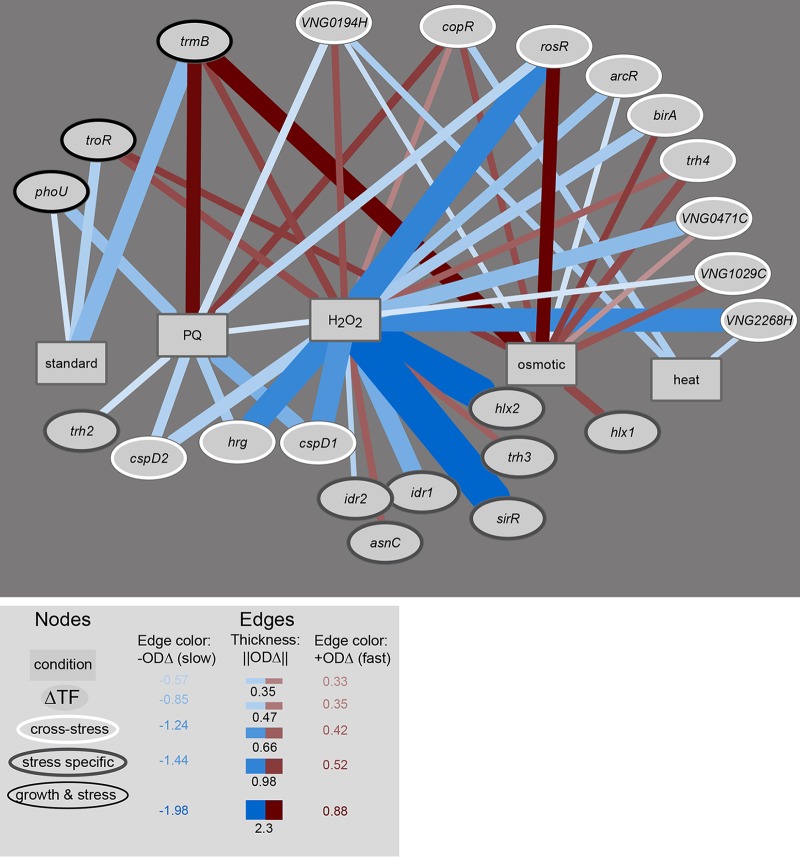
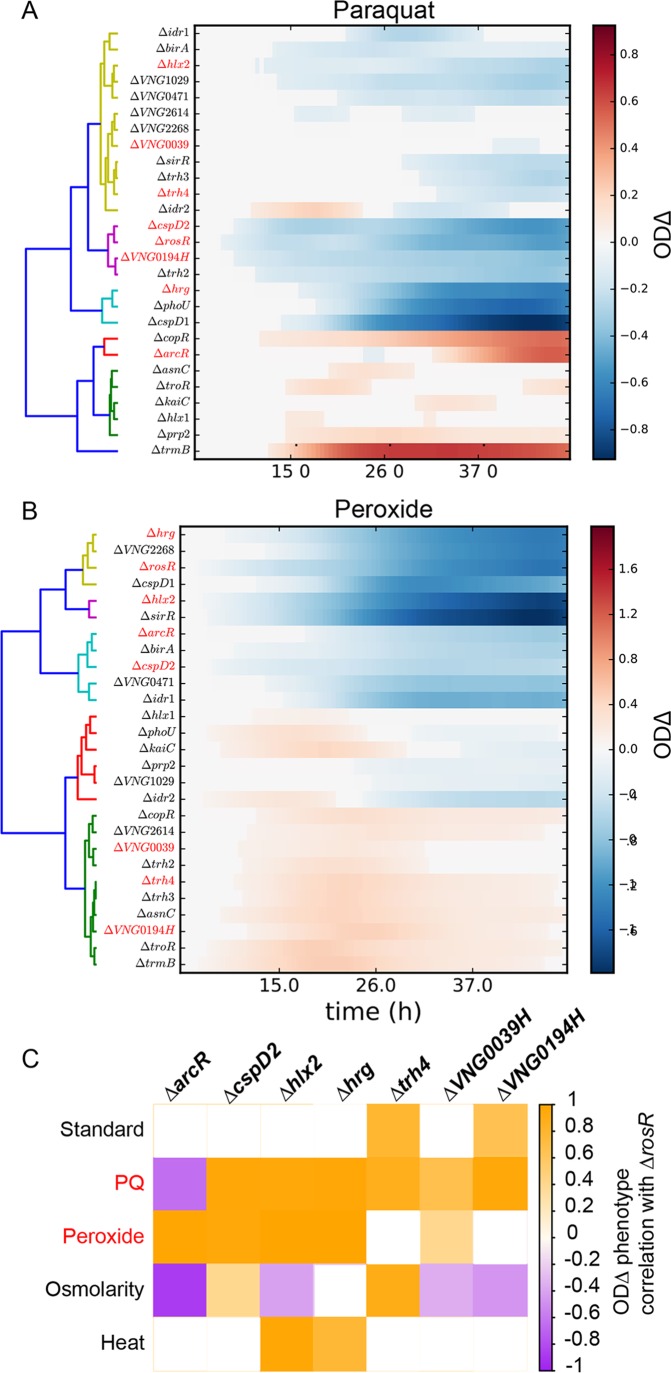
Gene regulatory networks (GRNs) are critical for dynamic transcriptional responses to environmental stress. However, the mechanisms by which GRN regulation adjusts physiology to enable stress survival remain unclear. Here we investigate the functions of transcription factors (TFs) within the global GRN of the stress-tolerant archaeal microorganism Halobacterium salinarum. We measured growth phenotypes of a panel of TF deletion mutants in high temporal resolution under heat shock, oxidative stress, and low-salinity conditions. To quantitate the noncanonical functional forms of the growth trajectories observed for these mutants, we developed a novel modeling framework based on Gaussian process regression and functional analysis of variance (FANOVA). We employ unique statistical tests to determine the significance of differential growth relative to the growth of the control strain. This analysis recapitulated known TF functions, revealed novel functions, and identified surprising secondary functions for characterized TFs. Strikingly, we observed that the majority of the TFs studied were required for growth under multiple stress conditions, pinpointing regulatory connections between the conditions tested. Correlations between quantitative phenotype trajectories of mutants are predictive of TF-TF connections within the GRN. These phenotypes are strongly concordant with predictions from statistical GRN models inferred from gene expression data alone. With genome-wide and targeted data sets, we provide detailed functional validation of novel TFs required for extreme oxidative stress and heat shock survival. Together, results presented in this study suggest that many TFs function under multiple conditions, thereby revealing high interconnectivity within the GRN and identifying the specific TFs required for communication between networks responding to disparate stressors. IMPORTANCE To ensure survival in the face of stress, microorganisms employ inducible damage repair pathways regulated by extensive and complex gene networks. Many archaea, microorganisms of the third domain of life, persist under extremes of temperature, salinity, and pH and under other conditions. In order to understand the cause-effect relationships between the dynamic function of the stress network and ultimate physiological consequences, this study characterized the physiological role of nearly one-third of all regulatory proteins known as transcription factors (TFs) in an archaeal organism. Using a unique quantitative phenotyping approach, we discovered functions for many novel TFs and revealed important secondary functions for known TFs. Surprisingly, many TFs are required for resisting multiple stressors, suggesting cross-regulation of stress responses. Through extensive validation experiments, we map the physiological roles of these novel TFs in stress response back to their position in the regulatory network wiring. This study advances understanding of the mechanisms underlying how microorganisms resist extreme stress. Given the generality of the methods employed, we expect that this study will enable future studies on how regulatory networks adjust cellular physiology in a diversity of organisms.
Keywords: Archaea; functional ANOVA; phenomics; transcription factors.
Figures
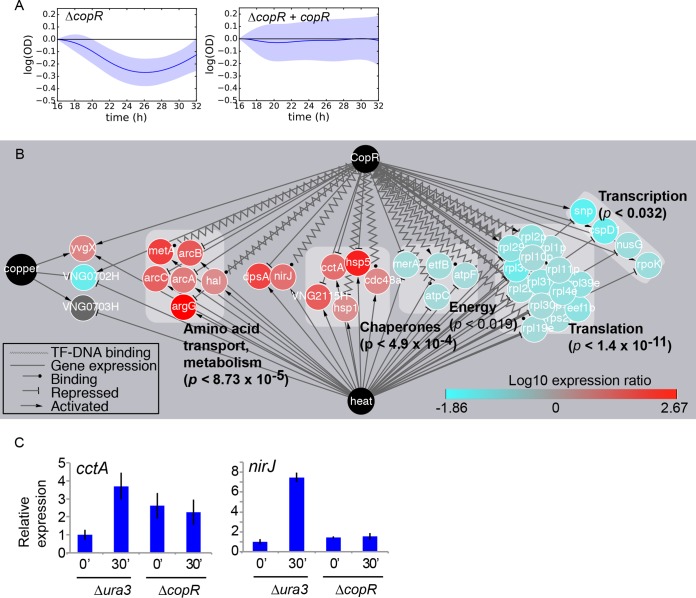
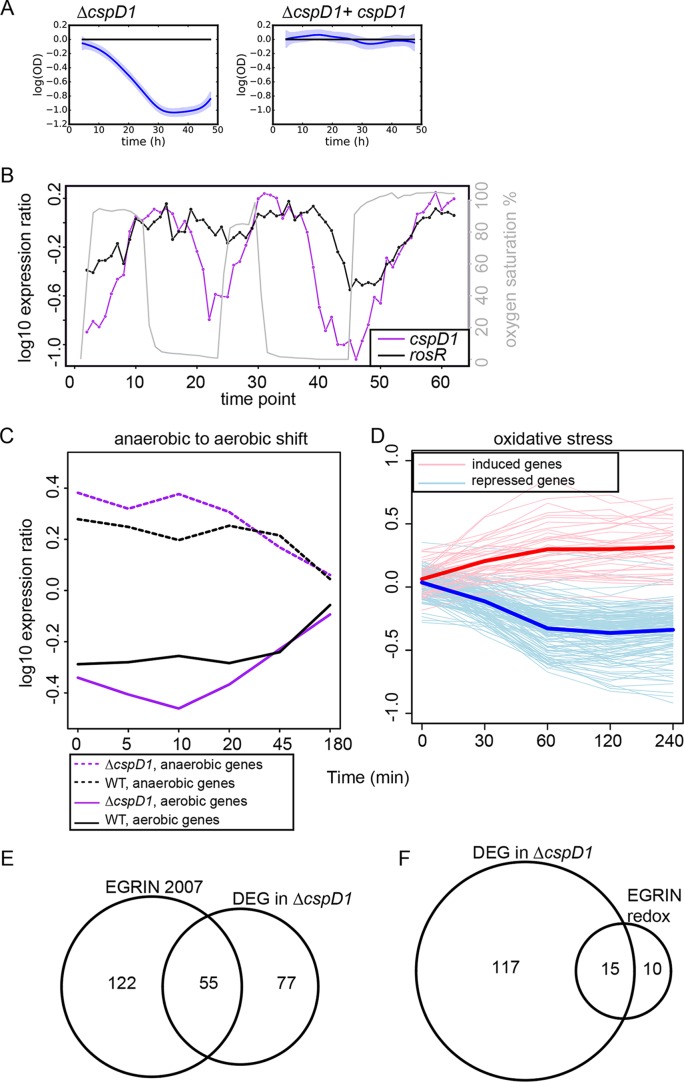
Similar articles
-
Systems biology approaches to defining transcription regulatory networks in halophilic archaea.Methods. 2015 Sep 15;86:102-14. doi: 10.1016/j.ymeth.2015.04.034. Epub 2015 May 12. Methods. 2015. PMID: 25976837
-
Transcriptional Regulation in Archaea: From Individual Genes to Global Regulatory Networks.Annu Rev Genet. 2017 Nov 27;51:143-170. doi: 10.1146/annurev-genet-120116-023413. Annu Rev Genet. 2017. PMID: 29178818 Review.
-
Network component analysis provides quantitative insights on an Arabidopsis transcription factor-gene regulatory network.BMC Syst Biol. 2013 Nov 14;7:126. doi: 10.1186/1752-0509-7-126. BMC Syst Biol. 2013. PMID: 24228871 Free PMC article.
-
Distinct tissue-specific transcriptional regulation revealed by gene regulatory networks in maize.BMC Plant Biol. 2018 Jun 7;18(1):111. doi: 10.1186/s12870-018-1329-y. BMC Plant Biol. 2018. PMID: 29879919 Free PMC article.
-
New insights on gene regulation in archaea.Comput Biol Chem. 2011 Dec 14;35(6):341-6. doi: 10.1016/j.compbiolchem.2011.10.006. Epub 2011 Oct 17. Comput Biol Chem. 2011. PMID: 22099630 Review.
Cited by
-
Navigating the archaeal frontier: insights and projections from bioinformatic pipelines.Front Microbiol. 2024 Sep 23;15:1433224. doi: 10.3389/fmicb.2024.1433224. eCollection 2024. Front Microbiol. 2024. PMID: 39380680 Free PMC article.
-
Genomic re-sequencing reveals mutational divergence across genetically engineered strains of model archaea.mSystems. 2025 Feb 18;10(2):e0108424. doi: 10.1128/msystems.01084-24. Epub 2025 Jan 10. mSystems. 2025. PMID: 39791890 Free PMC article.
-
Agl28 and Agl29 are key components of a Halobacterium salinarum N-glycosylation pathway.FEMS Microbiol Lett. 2023 Jan 17;370:fnad017. doi: 10.1093/femsle/fnad017. FEMS Microbiol Lett. 2023. PMID: 36866517 Free PMC article.
-
Stochasticity in transcriptional expression of a negative regulator of Arabidopsis ABA network.3 Biotech. 2019 Jan;9(1):15. doi: 10.1007/s13205-018-1542-2. Epub 2019 Jan 2. 3 Biotech. 2019. PMID: 30622853 Free PMC article.
-
Two different sulfotransferases modify sugars of the N-linked tetrasaccharide decorating Halobacterium salinarum glycoproteins.mBio. 2025 Apr 9;16(4):e0353424. doi: 10.1128/mbio.03534-24. Epub 2025 Feb 25. mBio. 2025. PMID: 39998273 Free PMC article.
References
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
