

The mitochondrion: a central architect of copper homeostasis
- PMID: 28952650
- PMCID: PMC5688007
- DOI: 10.1039/c7mt00221a
The mitochondrion: a central architect of copper homeostasis
Abstract
All known eukaryotes require copper for their development and survival. The essentiality of copper reflects its widespread use as a co-factor in conserved enzymes that catalyze biochemical reactions critical to energy production, free radical detoxification, collagen deposition, neurotransmitter biosynthesis and iron homeostasis. However, the prioritized use of copper poses an organism with a considerable challenge because, in its unbound form, copper can potentiate free radical production and displace iron-sulphur clusters to disrupt protein function. Protective mechanisms therefore evolved to mitigate this challenge and tightly regulate the acquisition, trafficking and storage of copper such that the metal ion is rarely found in its free form in the cell. Findings by a number of groups over the last ten years emphasize that this regulatory framework forms the foundation of a system that is capable of monitoring copper status and reprioritizing copper usage at both the cellular and systemic levels of organization. While the identification of relevant molecular mechanisms and signaling pathways has proven to be difficult and remains a barrier to our full understanding of the regulation of copper homeostasis, mounting evidence points to the mitochondrion as a pivotal hub in this regard in both healthy and diseased states. Here, we review our current understanding of copper handling pathways contained within the organelle and consider plausible mechanisms that may serve to functionally couple their activity to that of other cellular copper handling machinery to maintain copper homeostasis.
Figures
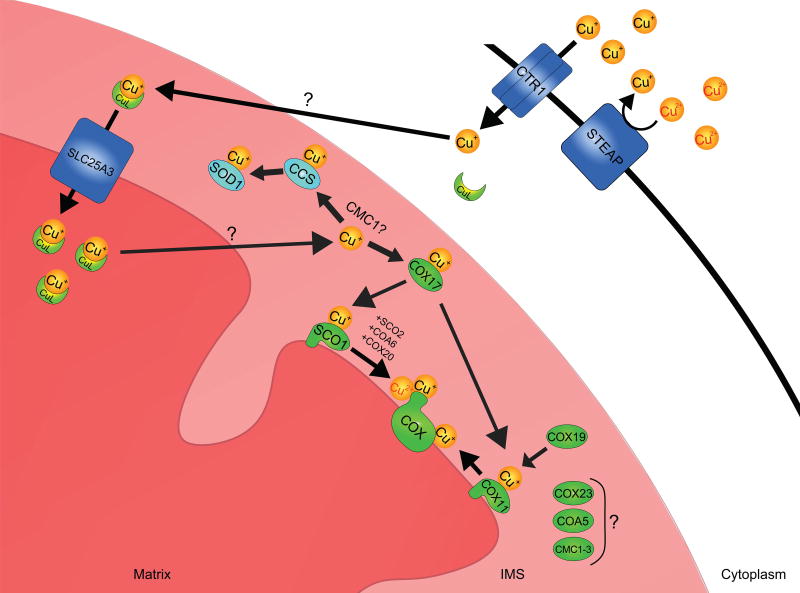
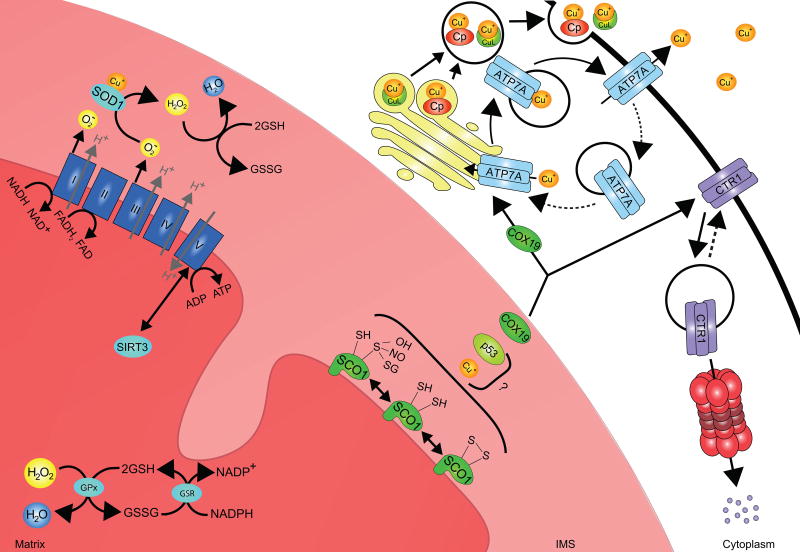
References
-
- Institute of Medicine (US) Panel on Micronutrients. Dietary Reference Intakes for Vitamin A, Vitamin K, Arsenic, Boron, Chromium, Copper, Iodine, Iron, Manganese, Molybdenum, Nickel, Silicon, Vanadium, and Zinc. National Academies Press (US); Washington (DC): 2001. pp. 224–257. - PubMed
-
- Prohaska JR. Impact of copper deficiency in humans. Ann. N.Y. Acad. Sci. 2014;1314:1–5. - PubMed
-
- Kim BE, Nevitt T, Thiele DJ. Mechanisms for copper acquisition, distribution and regulation. Nat. Chem. Biol. 2008;4:176–185. - PubMed
-
- Tisato F, Marzano C, Porchia M, Pellei M, Santini C. Copper in diseases treatments, and copper-based anticancer strategies. Med. Res. Rev. 2010;30:708–749. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
