

Incomplete dominance of deleterious alleles contributes substantially to trait variation and heterosis in maize
- PMID: 28953891
- PMCID: PMC5633198
- DOI: 10.1371/journal.pgen.1007019
Incomplete dominance of deleterious alleles contributes substantially to trait variation and heterosis in maize
Erratum in
-
Correction: Incomplete dominance of deleterious alleles contributes substantially to trait variation and heterosis in maize.PLoS Genet. 2021 Sep 28;17(9):e1009825. doi: 10.1371/journal.pgen.1009825. eCollection 2021 Sep. PLoS Genet. 2021. PMID: 34582434 Free PMC article.
Abstract
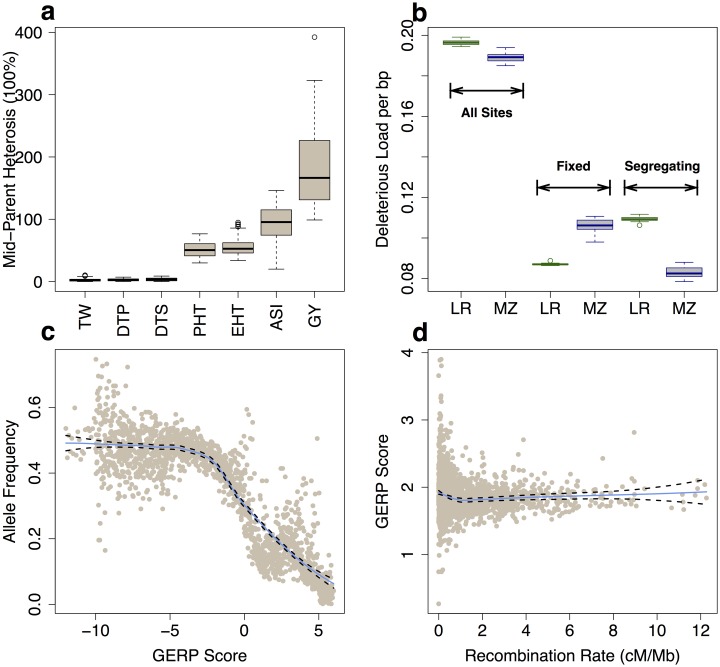
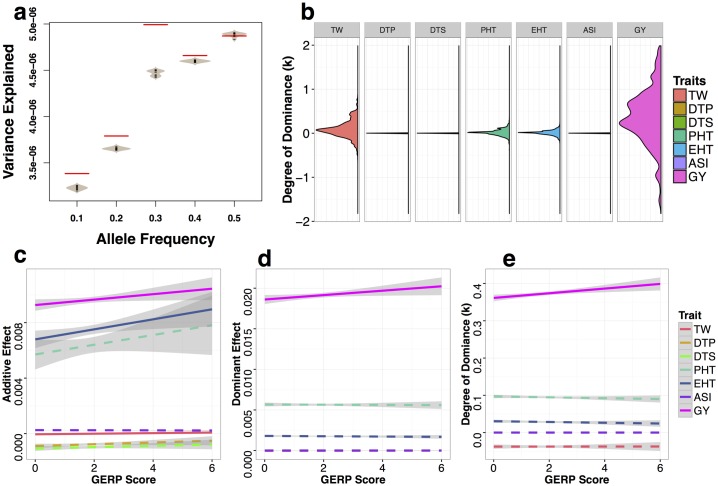
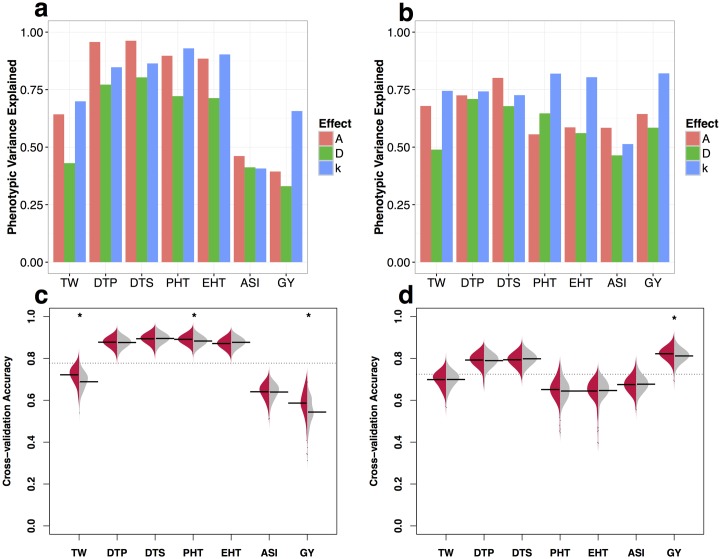
Deleterious alleles have long been proposed to play an important role in patterning phenotypic variation and are central to commonly held ideas explaining the hybrid vigor observed in the offspring of a cross between two inbred parents. We test these ideas using evolutionary measures of sequence conservation to ask whether incorporating information about putatively deleterious alleles can inform genomic selection (GS) models and improve phenotypic prediction. We measured a number of agronomic traits in both the inbred parents and hybrids of an elite maize partial diallel population and re-sequenced the parents of the population. Inbred elite maize lines vary for more than 350,000 putatively deleterious sites, but show a lower burden of such sites than a comparable set of traditional landraces. Our modeling reveals widespread evidence for incomplete dominance at these loci, and supports theoretical models that more damaging variants are usually more recessive. We identify haplotype blocks using an identity-by-decent (IBD) analysis and perform genomic prediction analyses in which we weigh blocks on the basis of complementation for segregating putatively deleterious variants. Cross-validation results show that incorporating sequence conservation in genomic selection improves prediction accuracy for grain yield and other fitness-related traits as well as heterosis for those traits. Our results provide empirical support for an important role for incomplete dominance of deleterious alleles in explaining heterosis and demonstrate the utility of incorporating functional annotation in phenotypic prediction and plant breeding.
Conflict of interest statement
I have read the journal’s policy and the authors of this manuscript have the following competing interests: Rita Mumm received research funding from Kellogg Company to support field work, equipment, and travel. She is a paid consultant for Mars Inc. and serves on the leadership team of the African Orphan Crop Consortium which is funded in part by Mars Inc. Jeffrey Ross-Ibarra received research funding from DuPont Pioneer and Mars Inc. Sofiane Mezmouk is a current employee of KWS. Andy Baumgarten is a current employee of DuPont Pioneer. All authors declare no additional competing interests, and none of the funders played any role in the study design; collection, analysis, and interpretation of data; writing of the paper; and/or decision to submit for publication.
Figures
References
-
- Mitchell-Olds T, Willis JH, Goldstein DB. Which evolutionary processes influence natural genetic variation for phenotypic traits? Nature Reviews Genetics. 2007;8(11):845–856. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
