

Predicting Oral Drug Absorption: Mini Review on Physiologically-Based Pharmacokinetic Models
- PMID: 28954416
- PMCID: PMC5750647
- DOI: 10.3390/pharmaceutics9040041
Predicting Oral Drug Absorption: Mini Review on Physiologically-Based Pharmacokinetic Models
Abstract
Most marketed drugs are administered orally, despite the complex process of oral absorption that is difficult to predict. Oral bioavailability is dependent on the interplay between many processes that are dependent on both compound and physiological properties. Because of this complexity, computational oral physiologically-based pharmacokinetic (PBPK) models have emerged as a tool to integrate these factors in an attempt to mechanistically capture the process of oral absorption. These models use inputs from in vitro assays to predict the pharmacokinetic behavior of drugs in the human body. The most common oral PBPK models are compartmental approaches, in which the gastrointestinal tract is characterized as a series of compartments through which the drug transits. The focus of this review is on the development of oral absorption PBPK models, followed by a brief discussion of the major applications of oral PBPK models in the pharmaceutical industry.
Keywords: food-effect; formulation simulation; oral absorption; pH effect; physiologically-based pharmacokinetic modeling.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
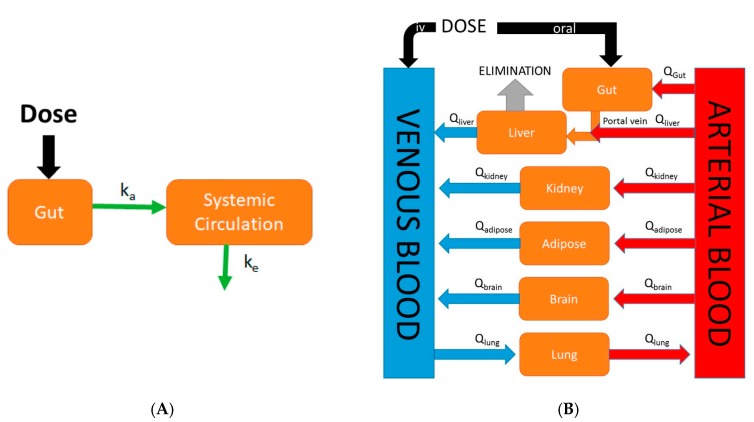
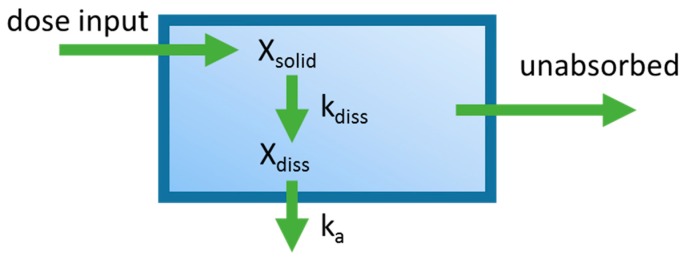
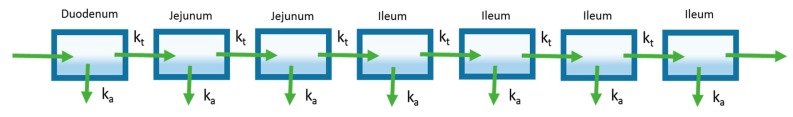
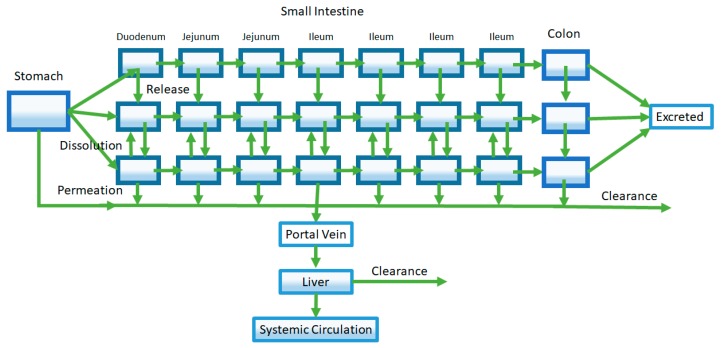
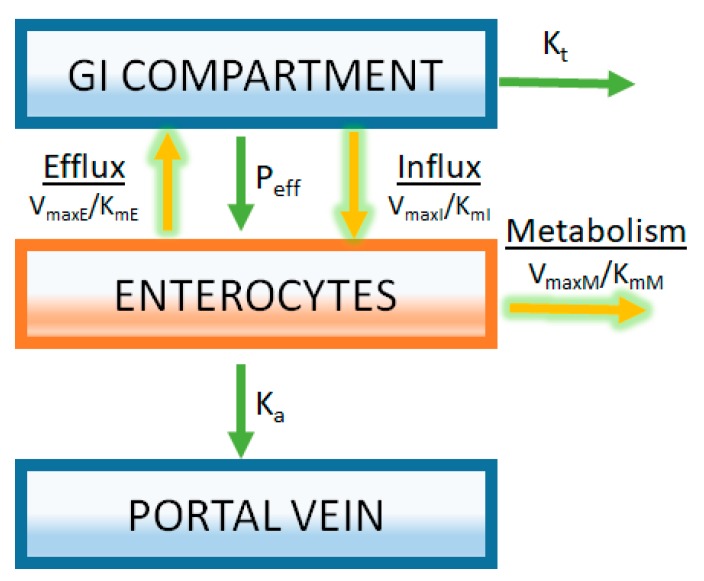
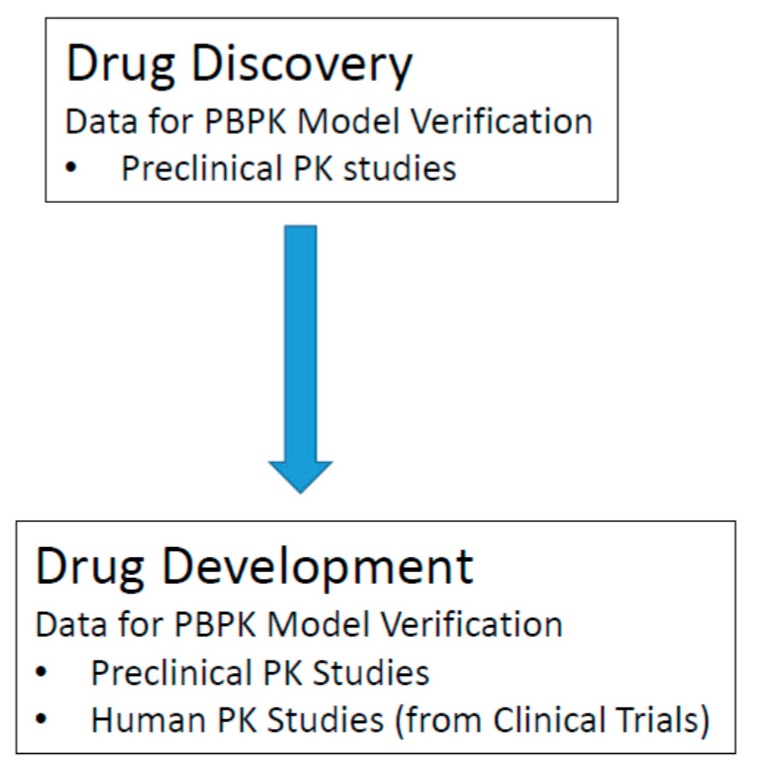
References
-
- Waring M.J., Arrowsmith J., Leach A.R., Leeson P.D., Mandrell S., Owen R.M., Pairaudeau G., Pennie W.D., Pickett S.D., Wang J., et al. An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nat. Rev. Drug Discov. 2015;14:475–486. doi: 10.1038/nrd4609. - DOI - PubMed
-
- Ward K.W. Reducing Drug Attrition. Springer; Berlin, Germany: 2012. Optimizing pharmacokinetic properties and attaining candidate selection; pp. 73–95. Topics in Medicinal Chemistry.
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
