

Necroptotic debris including damaged mitochondria elicits sepsis-like syndrome during late-phase tularemia
- PMID: 28955505
- PMCID: PMC5611684
- DOI: 10.1038/cddiscovery.2017.56
Necroptotic debris including damaged mitochondria elicits sepsis-like syndrome during late-phase tularemia
Abstract
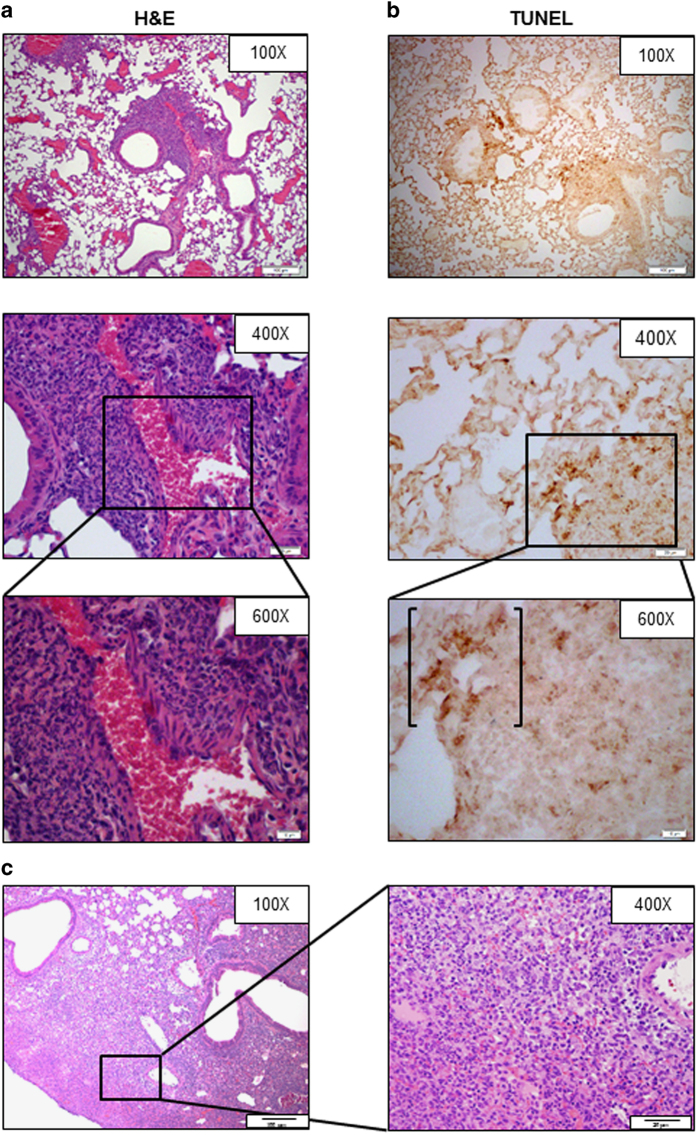
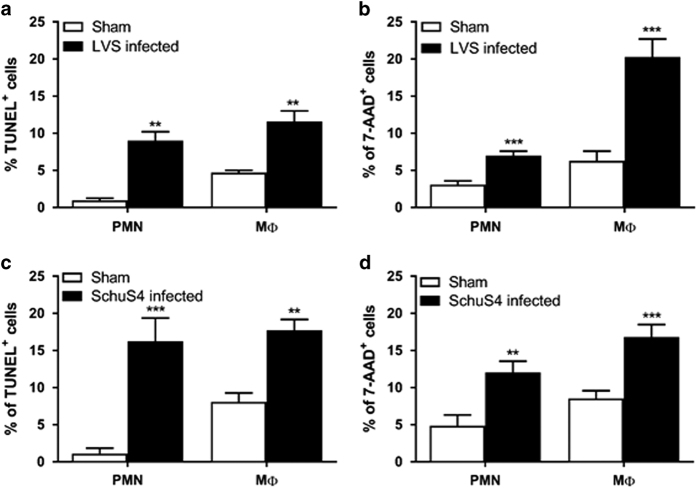
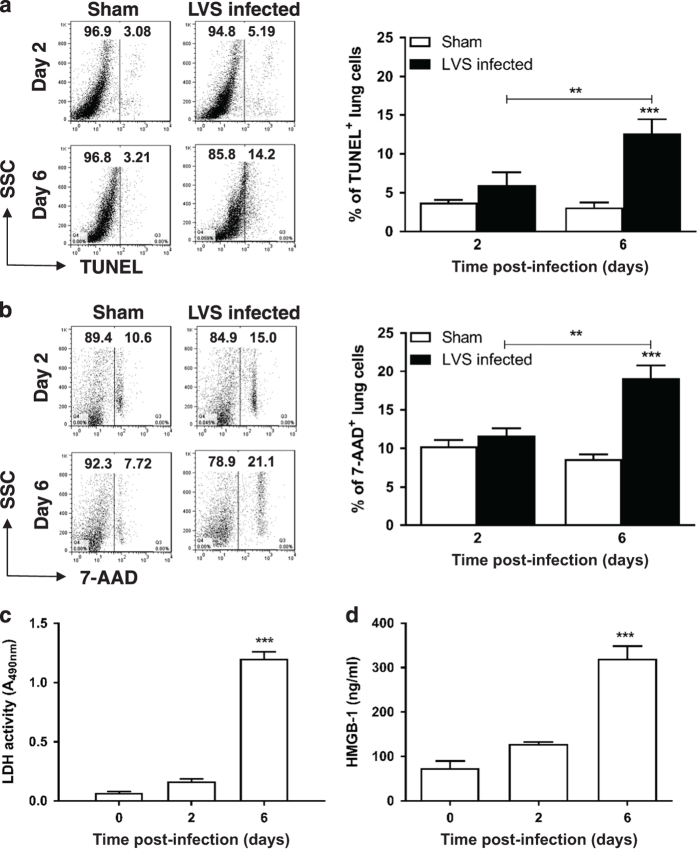
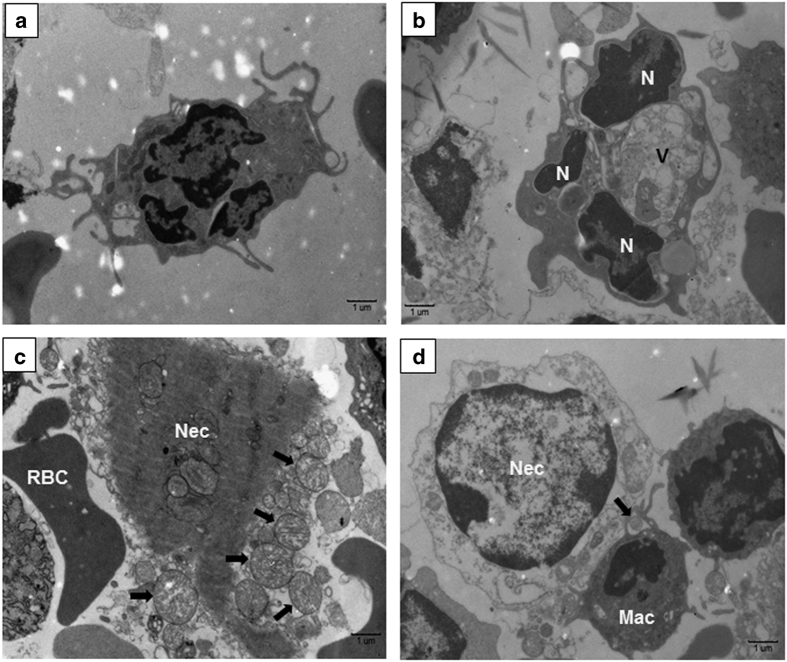
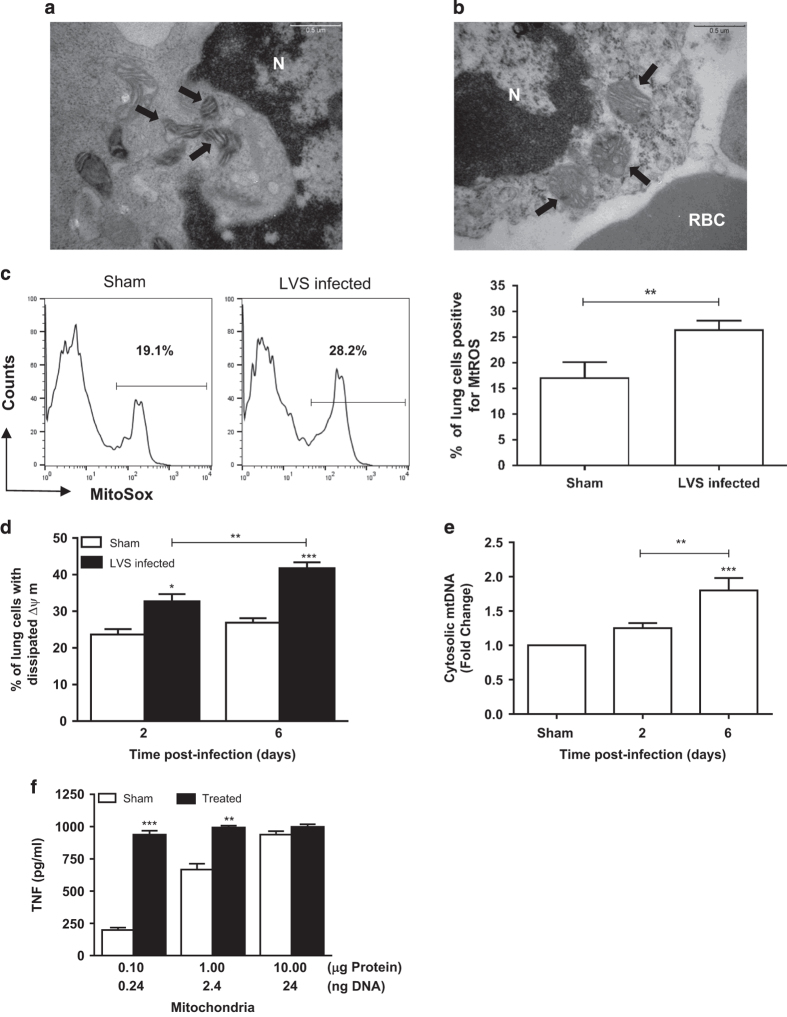
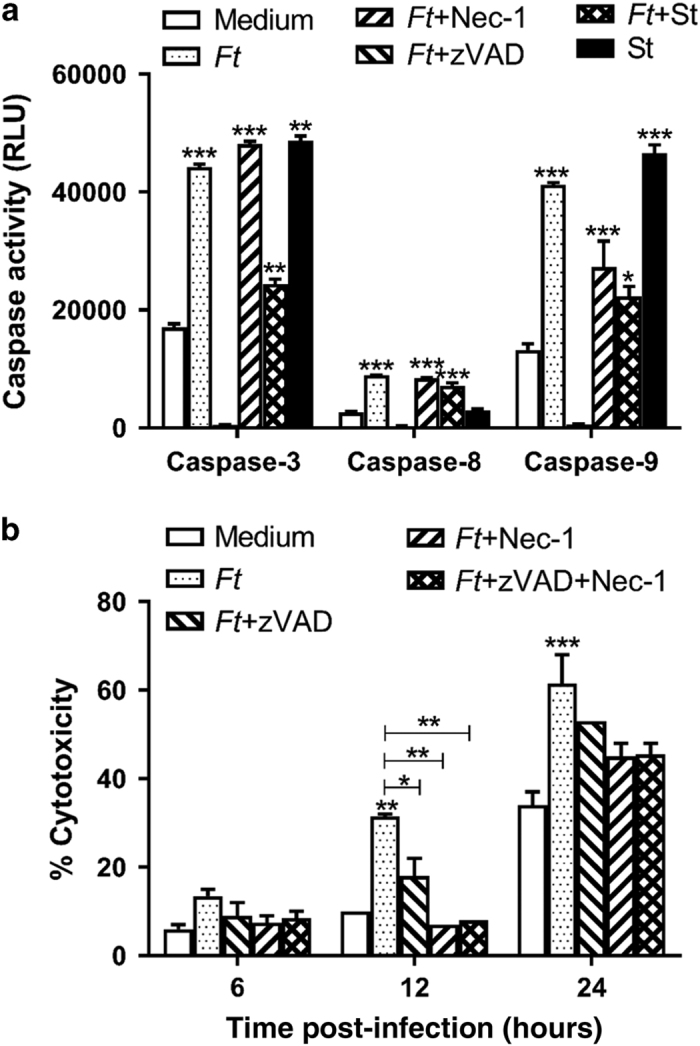
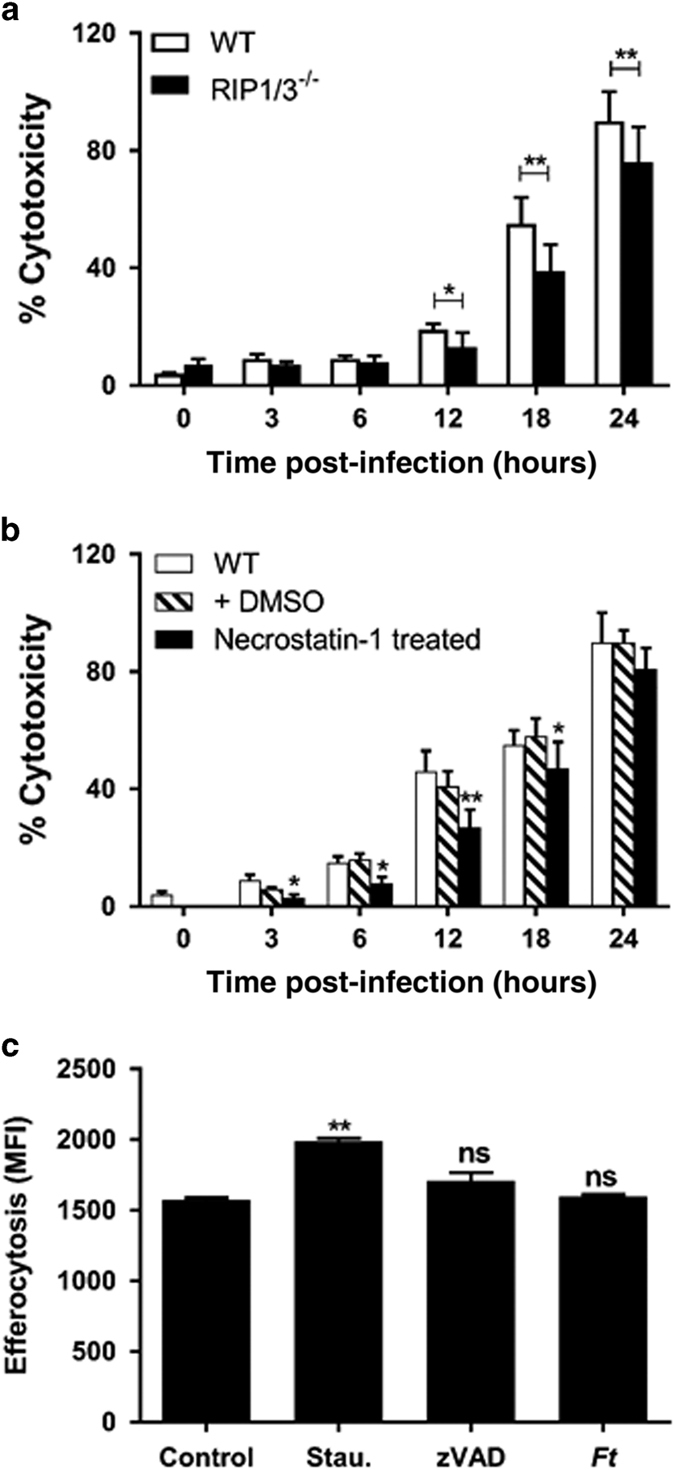
Infection with Francisella tularensis ssp. tularensis (Ft) strain SchuS4 causes an often lethal disease known as tularemia in rodents, non-human primates, and humans. Ft subverts host cell death programs to facilitate their exponential replication within macrophages and other cell types during early respiratory infection (⩽72 h). The mechanism(s) by which cell death is triggered remains incompletely defined, as does the impact of Ft on mitochondria, the host cell's organellar 'canary in a coal mine'. Herein, we reveal that Ft infection of host cells, particularly macrophages and polymorphonuclear leukocytes, drives necroptosis via a receptor-interacting protein kinase 1/3-mediated mechanism. During necroptosis mitochondria and other organelles become damaged. Ft-induced mitochondrial damage is characterized by: (i) a decrease in membrane potential and consequent mitochondrial oncosis or swelling, (ii) increased generation of superoxide radicals, and (iii) release of intact or damaged mitochondria into the lung parenchyma. Host cell recognition of and response to released mitochondria and other damage-associated molecular patterns engenders a sepsis-like syndrome typified by production of TNF, IL-1β, IL-6, IL-12p70, and IFN-γ during late-phase tularemia (⩾72 h), but are absent early during infection.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Inflammasome-Independent NLRP3 Restriction of a Protective Early Neutrophil Response to Pulmonary Tularemia.PLoS Pathog. 2016 Dec 7;12(12):e1006059. doi: 10.1371/journal.ppat.1006059. eCollection 2016 Dec. PLoS Pathog. 2016. PMID: 27926940 Free PMC article.
-
An Immature Myeloid/Myeloid-Suppressor Cell Response Associated with Necrotizing Inflammation Mediates Lethal Pulmonary Tularemia.PLoS Pathog. 2016 Mar 25;12(3):e1005517. doi: 10.1371/journal.ppat.1005517. eCollection 2016 Mar. PLoS Pathog. 2016. PMID: 27015566 Free PMC article.
-
Lipoxin A4, a 5-lipoxygenase pathway metabolite, modulates immune response during acute respiratory tularemia.J Leukoc Biol. 2017 Feb;101(2):531-542. doi: 10.1189/jlb.4A0815-365RR. Epub 2016 Sep 14. J Leukoc Biol. 2017. PMID: 27630217 Free PMC article.
-
Vaccination evokes gender-dependent protection against tularemia infection in C57BL/6Tac mice.Vaccine. 2016 Jun 17;34(29):3396-404. doi: 10.1016/j.vaccine.2016.04.054. Epub 2016 May 13. Vaccine. 2016. PMID: 27182819 Free PMC article.
-
Francisella tularensis: activation of the inflammasome.Ann N Y Acad Sci. 2007 Jun;1105:219-37. doi: 10.1196/annals.1409.005. Epub 2007 Mar 29. Ann N Y Acad Sci. 2007. PMID: 17395724 Review.
Cited by
-
Necroptosis in bacterial infections.Front Immunol. 2024 Jun 12;15:1394857. doi: 10.3389/fimmu.2024.1394857. eCollection 2024. Front Immunol. 2024. PMID: 38933265 Free PMC article. Review.
-
Mitochondria Released by Apoptotic Cell Death Initiate Innate Immune Responses.Immunohorizons. 2018 Dec;2(11):384-397. doi: 10.4049/immunohorizons.1800063. Immunohorizons. 2018. PMID: 30847435 Free PMC article.
-
Tularemia - a re-emerging disease with growing concern.Vet Q. 2023 Dec;43(1):1-16. doi: 10.1080/01652176.2023.2277753. Epub 2023 Nov 18. Vet Q. 2023. PMID: 37916743 Free PMC article.
-
PANoptosis in microbial infection.Curr Opin Microbiol. 2021 Feb;59:42-49. doi: 10.1016/j.mib.2020.07.012. Epub 2020 Aug 20. Curr Opin Microbiol. 2021. PMID: 32829024 Free PMC article. Review.
-
Multifaceted Roles of Mitochondrial Components and Metabolites in Metabolic Diseases and Cancer.Int J Mol Sci. 2020 Jun 20;21(12):4405. doi: 10.3390/ijms21124405. Int J Mol Sci. 2020. PMID: 32575796 Free PMC article. Review.
References
-
- Cui Z, Song E, Hu DN, Chen M, Rosen R, McCormick SA. Butein-induces apoptosis in human uveal melanoma cells through mitochondrial apoptosis pathway. Curr Eye Res 2012; 37: 730–739. - PubMed
-
- Karin M, Lin A. NF-[kappa]B at the crossroads of life and death. Nat Immunol 2002; 3: 221–227. - PubMed
-
- Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol 2008; 9: 231–241. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
