

Folding Membrane Proteins by Deep Transfer Learning
- PMID: 28957654
- PMCID: PMC5637520
- DOI: 10.1016/j.cels.2017.09.001
Folding Membrane Proteins by Deep Transfer Learning
Abstract
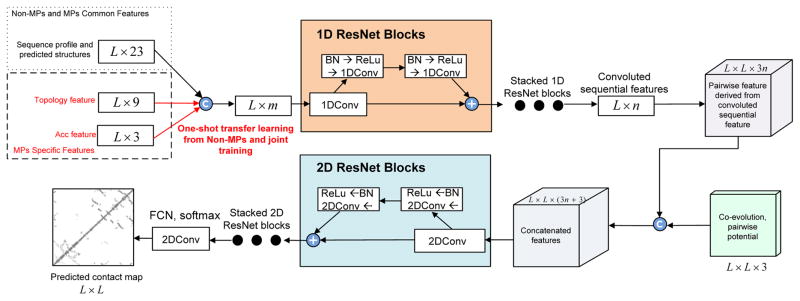
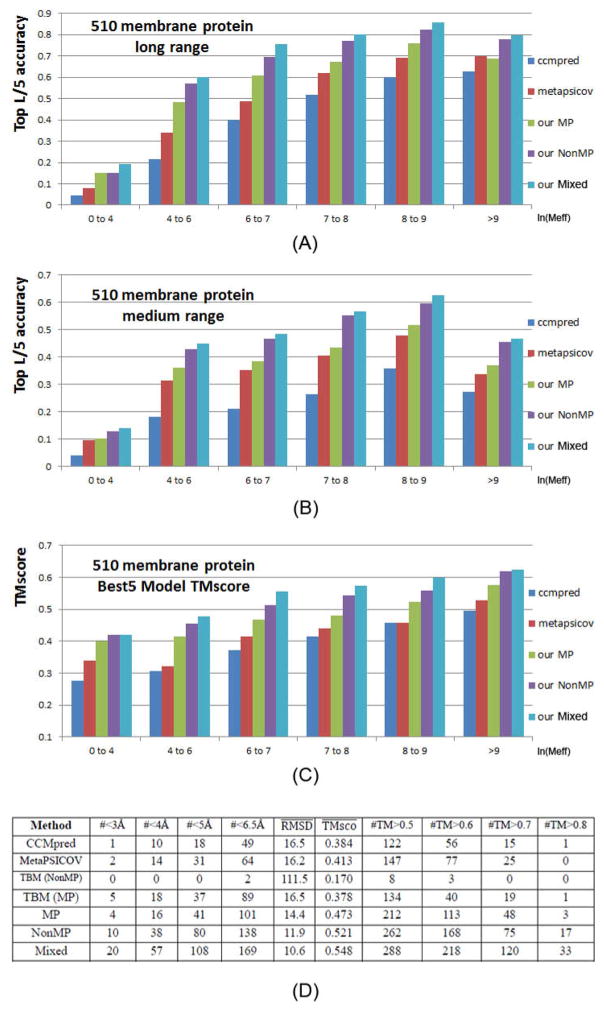
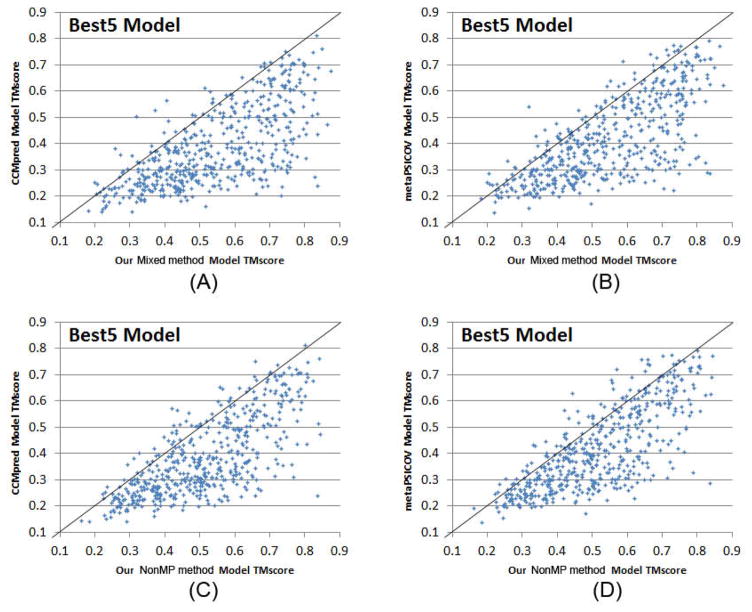
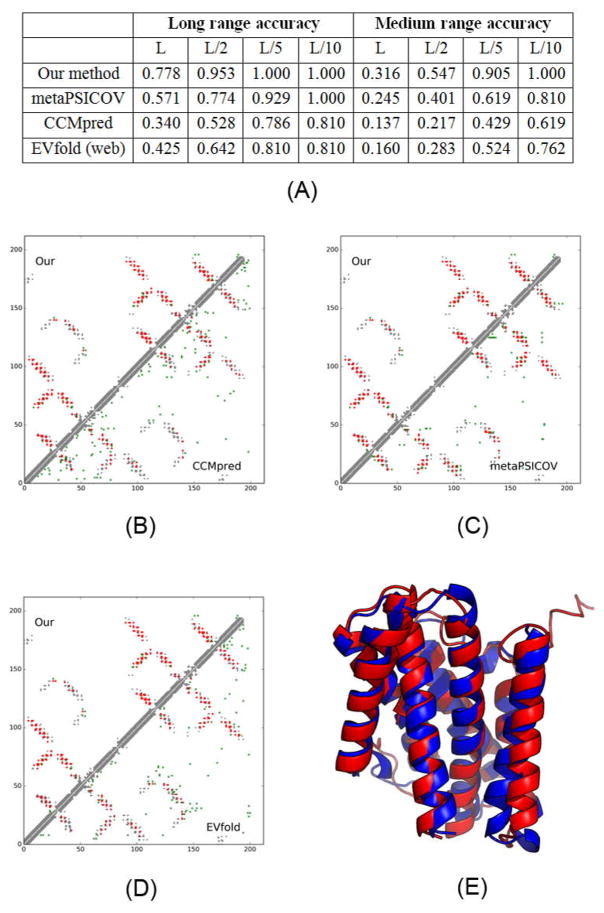
Computational elucidation of membrane protein (MP) structures is challenging partially due to lack of sufficient solved structures for homology modeling. Here, we describe a high-throughput deep transfer learning method that first predicts MP contacts by learning from non-MPs and then predicts 3D structure models using the predicted contacts as distance restraints. Tested on 510 non-redundant MPs, our method has contact prediction accuracy at least 0.18 better than existing methods, predicts correct folds for 218 MPs, and generates 3D models with root-mean-square deviation (RMSD) less than 4 and 5 Å for 57 and 108 MPs, respectively. A rigorous blind test in the continuous automated model evaluation project shows that our method predicted high-resolution 3D models for two recent test MPs of 210 residues with RMSD ∼2 Å. We estimated that our method could predict correct folds for 1,345-1,871 reviewed human multi-pass MPs including a few hundred new folds, which shall facilitate the discovery of drugs targeting at MPs.
Keywords: co-evolution analysis; deep learning; deep transfer learning; homology modeling; membrane protein contact prediction; membrane protein folding; multiple sequence alignment.
Copyright © 2017 Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Accurate De Novo Prediction of Protein Contact Map by Ultra-Deep Learning Model.PLoS Comput Biol. 2017 Jan 5;13(1):e1005324. doi: 10.1371/journal.pcbi.1005324. eCollection 2017 Jan. PLoS Comput Biol. 2017. PMID: 28056090 Free PMC article.
-
PredMP: a web server for de novo prediction and visualization of membrane proteins.Bioinformatics. 2019 Feb 15;35(4):691-693. doi: 10.1093/bioinformatics/bty684. Bioinformatics. 2019. PMID: 30084960 Free PMC article.
-
Challenges of Protein-Protein Docking of the Membrane Proteins.Methods Mol Biol. 2024;2780:203-255. doi: 10.1007/978-1-0716-3985-6_12. Methods Mol Biol. 2024. PMID: 38987471
-
Computational modeling of membrane proteins.Proteins. 2015 Jan;83(1):1-24. doi: 10.1002/prot.24703. Epub 2014 Nov 19. Proteins. 2015. PMID: 25355688 Free PMC article. Review.
-
Membrane proteins structures: A review on computational modeling tools.Biochim Biophys Acta Biomembr. 2017 Oct;1859(10):2021-2039. doi: 10.1016/j.bbamem.2017.07.008. Epub 2017 Jul 15. Biochim Biophys Acta Biomembr. 2017. PMID: 28716627 Review.
Cited by
-
Prokaryotic and eukaryotic promoters identification based on residual network transfer learning.Bioprocess Biosyst Eng. 2022 May;45(5):955-967. doi: 10.1007/s00449-022-02716-w. Epub 2022 Mar 13. Bioprocess Biosyst Eng. 2022. PMID: 35279747
-
Identification of residue pairing in interacting β-strands from a predicted residue contact map.BMC Bioinformatics. 2018 Apr 19;19(1):146. doi: 10.1186/s12859-018-2150-1. BMC Bioinformatics. 2018. PMID: 29673311 Free PMC article.
-
Machine learning in computational modelling of membrane protein sequences and structures: From methodologies to applications.Comput Struct Biotechnol J. 2023 Jan 28;21:1205-1226. doi: 10.1016/j.csbj.2023.01.036. eCollection 2023. Comput Struct Biotechnol J. 2023. PMID: 36817959 Free PMC article. Review.
-
Structure of the human BBSome core complex.Elife. 2020 Jan 17;9:e53910. doi: 10.7554/eLife.53910. Elife. 2020. PMID: 31951201 Free PMC article.
-
Computational-approach understanding the structure-function prophecy of Fibrinolytic Protease RFEA1 from Bacillus cereus RSA1.PeerJ. 2021 Jun 4;9:e11570. doi: 10.7717/peerj.11570. eCollection 2021. PeerJ. 2021. PMID: 34141495 Free PMC article.
References
-
- BRIINGER AT, ADAMS PD, CLORE GM, DELANO WL, GROS P, GROSSE-KUNSTLEVE RW, JIANG JS, KUSZEWSKI J, NILGES M, PANNU NS. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
