

Epigenetic restriction of extraembryonic lineages mirrors the somatic transition to cancer
- PMID: 28959968
- PMCID: PMC5789792
- DOI: 10.1038/nature23891
Epigenetic restriction of extraembryonic lineages mirrors the somatic transition to cancer
Abstract
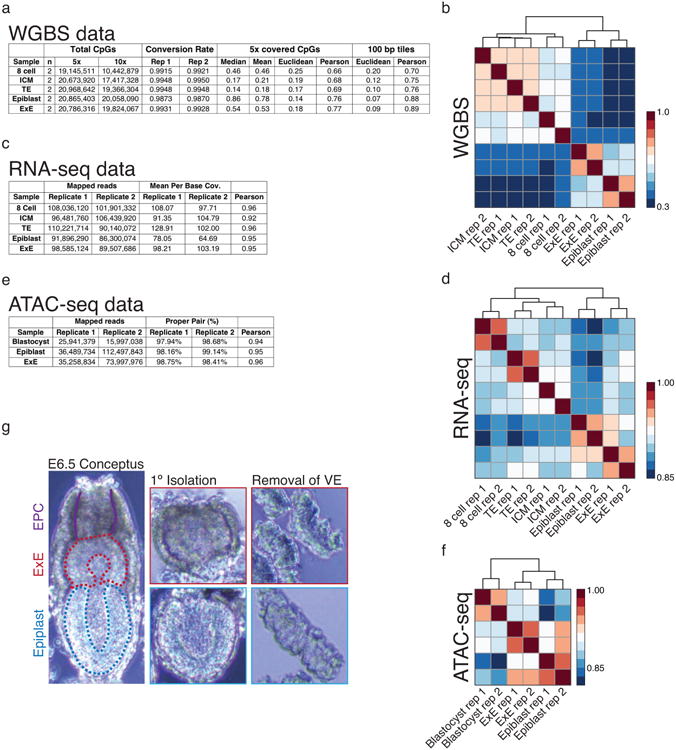
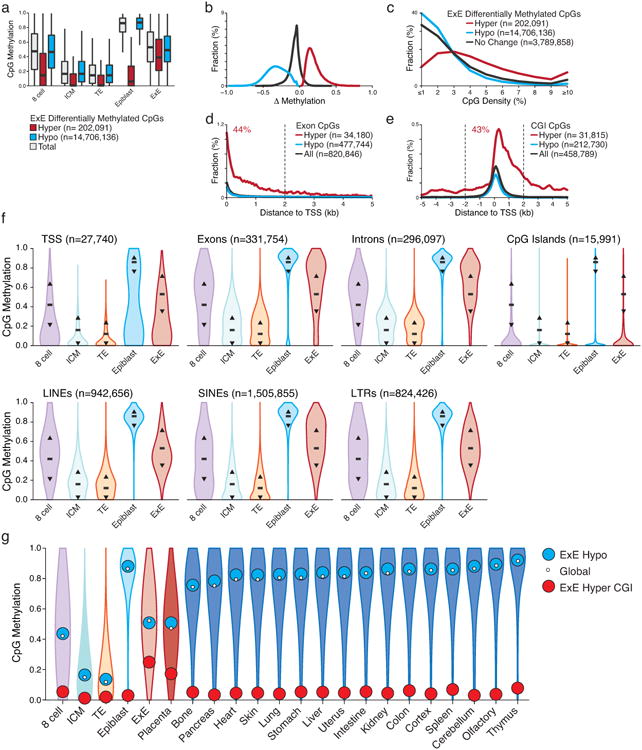
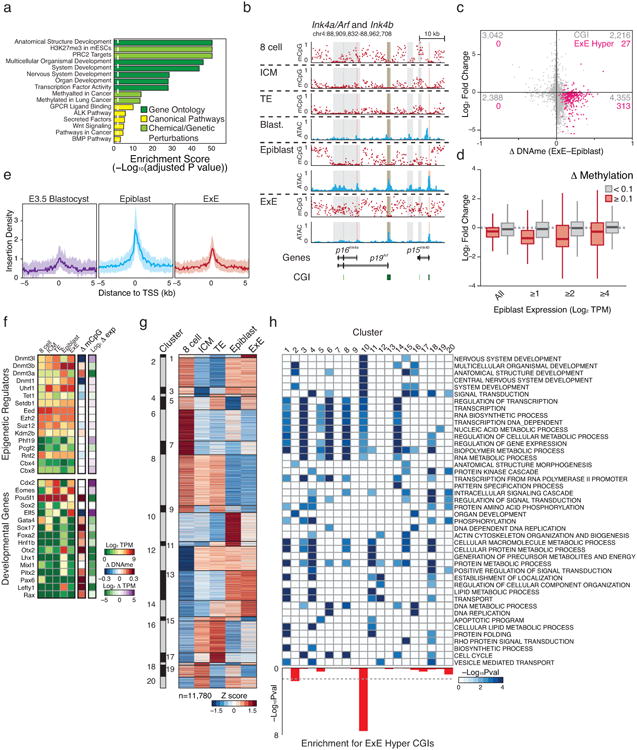
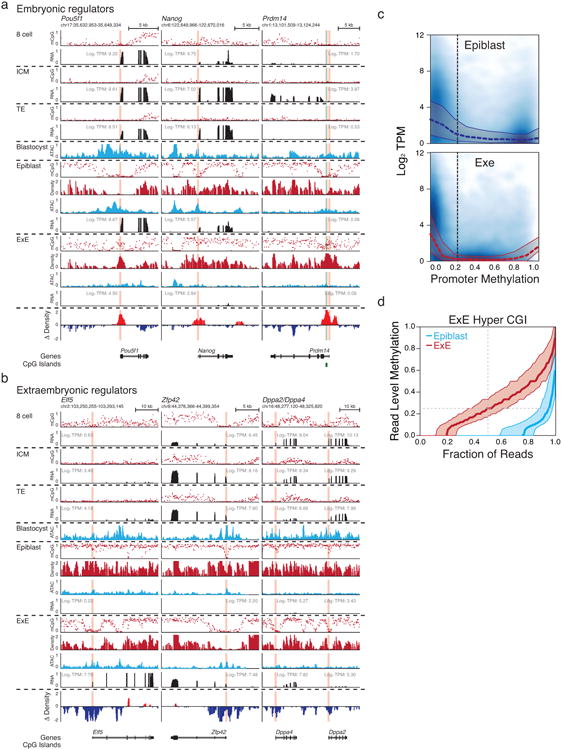
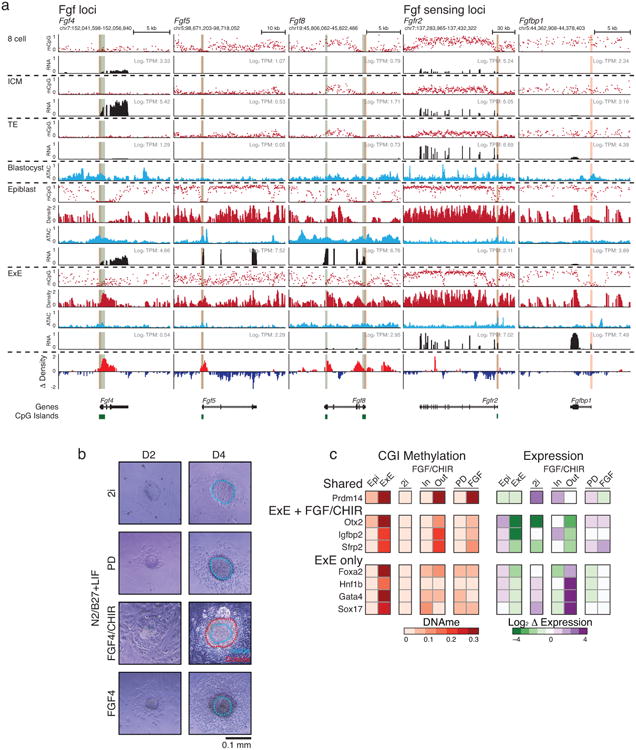
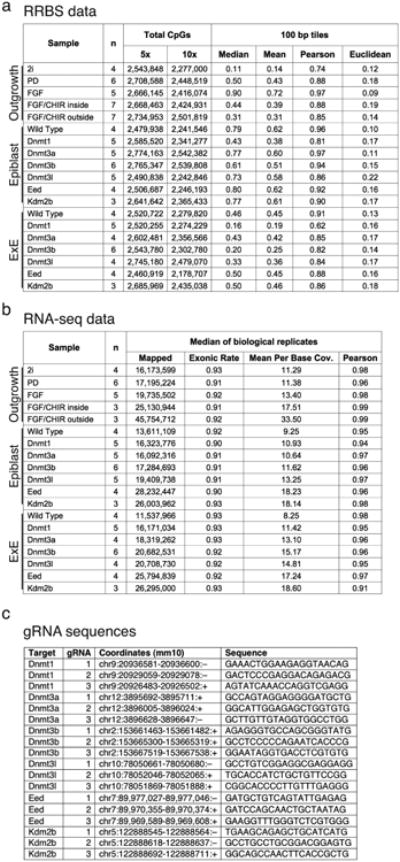
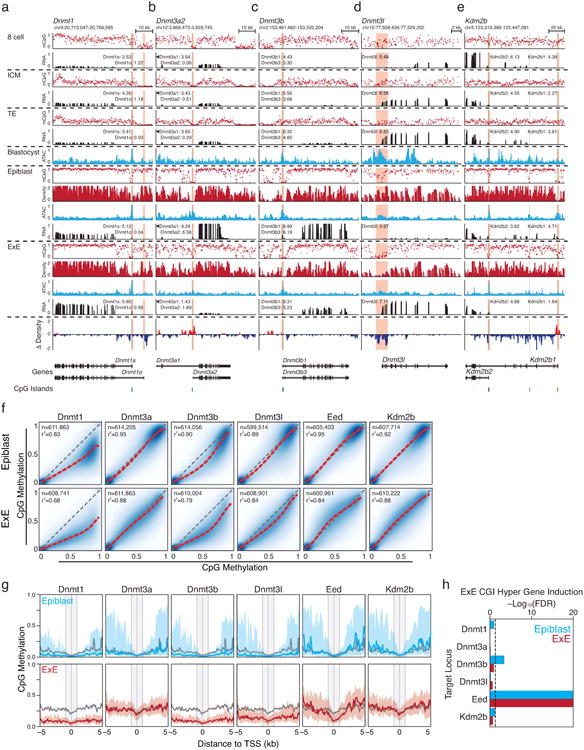
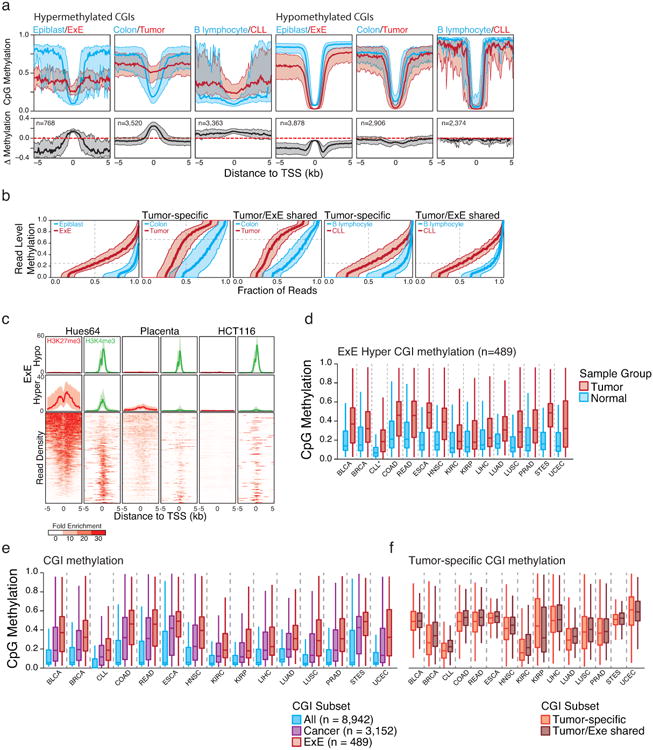
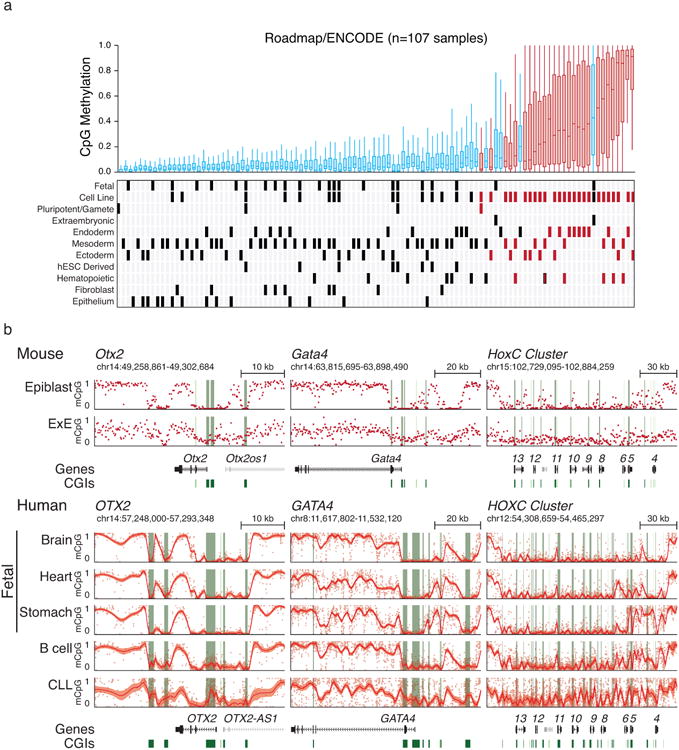
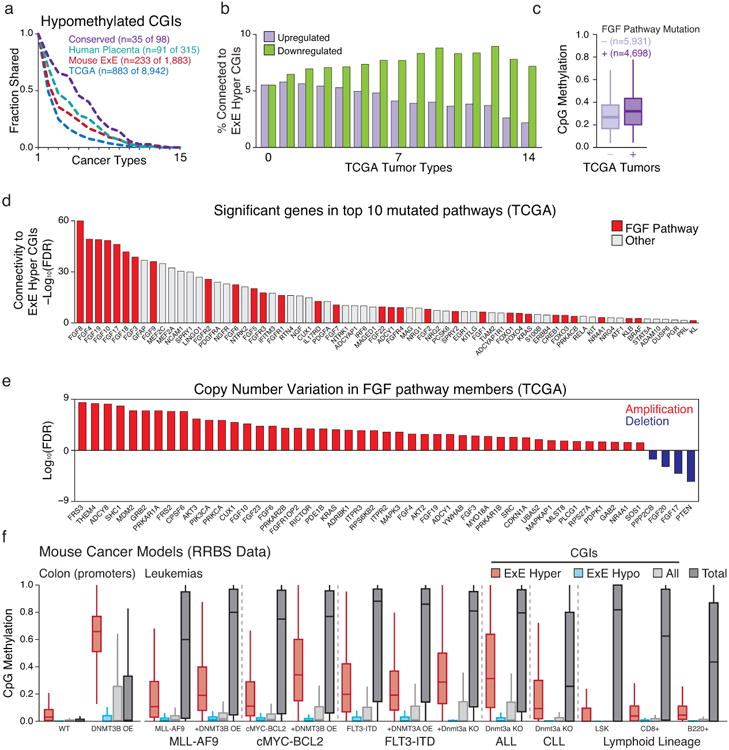
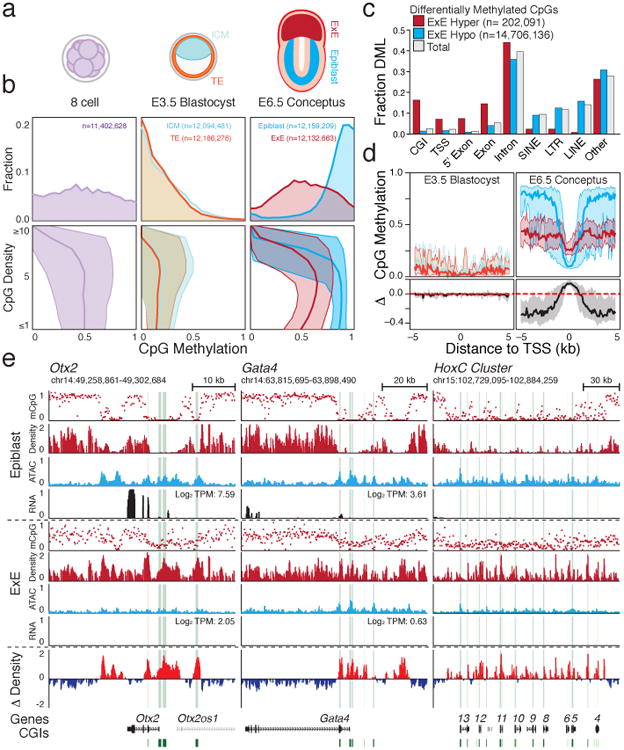
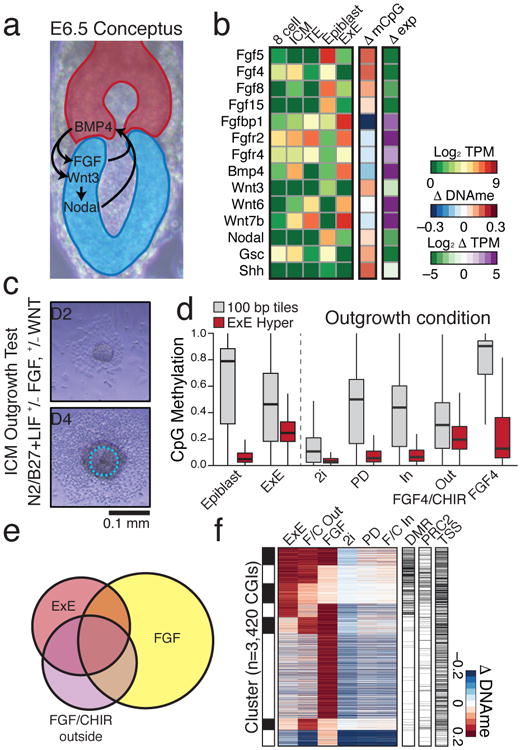
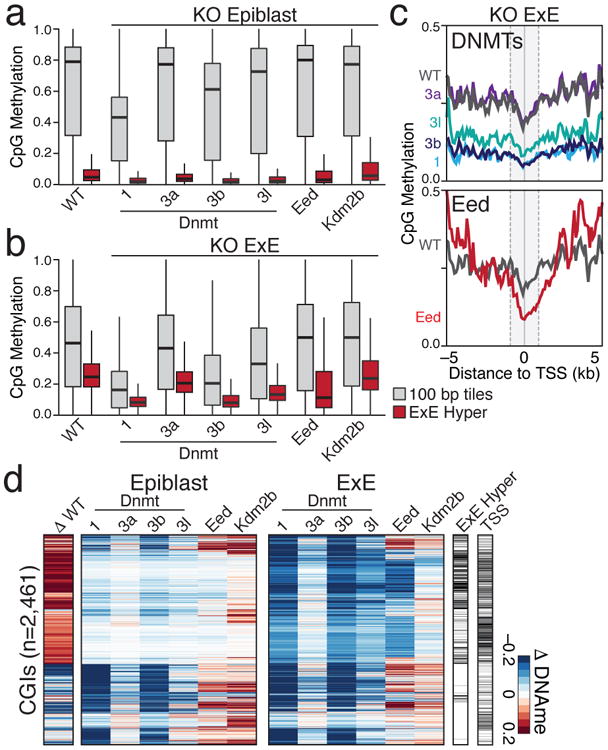
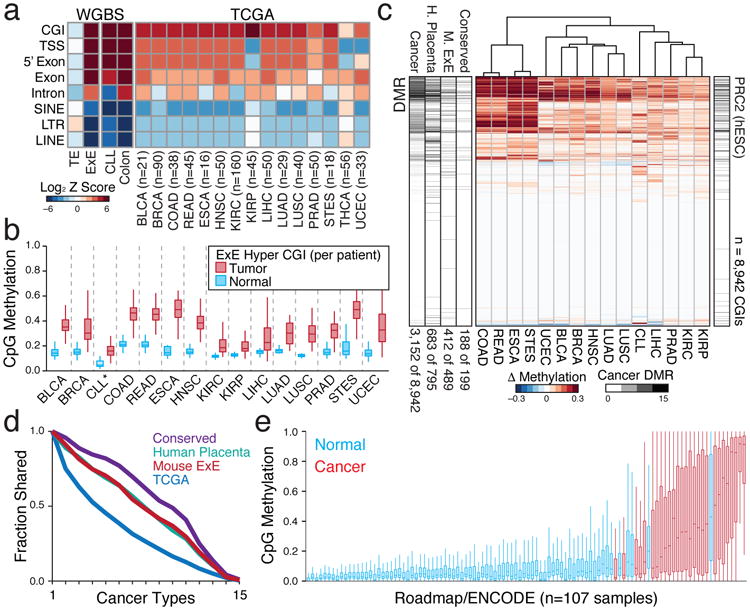
In mammals, the canonical somatic DNA methylation landscape is established upon specification of the embryo proper and subsequently disrupted within many cancer types. However, the underlying mechanisms that direct this genome-scale transformation remain elusive, with no clear model for its systematic acquisition or potential developmental utility. Here, we analysed global remethylation from the mouse preimplantation embryo into the early epiblast and extraembryonic ectoderm. We show that these two states acquire highly divergent genomic distributions with substantial disruption of bimodal, CpG density-dependent methylation in the placental progenitor. The extraembryonic epigenome includes specific de novo methylation at hundreds of embryonically protected CpG island promoters, particularly those that are associated with key developmental regulators and are orthologously methylated across most human cancer types. Our data suggest that the evolutionary innovation of extraembryonic tissues may have required co-option of DNA methylation-based suppression as an alternative to regulation by Polycomb-group proteins, which coordinate embryonic germ-layer formation in response to extraembryonic cues. Moreover, we establish that this decision is made deterministically, downstream of promiscuously used-and frequently oncogenic-signalling pathways, via a novel combination of epigenetic cofactors. Methylation of developmental gene promoters during tumorigenesis may therefore reflect the misappropriation of an innate trajectory and the spontaneous reacquisition of a latent, developmentally encoded epigenetic landscape.
Figures
Comment in
-
Evidence for Converging DNA Methylation Pathways in Placenta and Cancer.Dev Cell. 2017 Nov 6;43(3):257-258. doi: 10.1016/j.devcel.2017.10.009. Dev Cell. 2017. PMID: 29112847
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
