

Mimicking Intermolecular Interactions of Tight Protein-Protein Complexes for Small-Molecule Antagonists
- PMID: 28960868
- PMCID: PMC6067920
- DOI: 10.1002/cmdc.201700572
Mimicking Intermolecular Interactions of Tight Protein-Protein Complexes for Small-Molecule Antagonists
Abstract
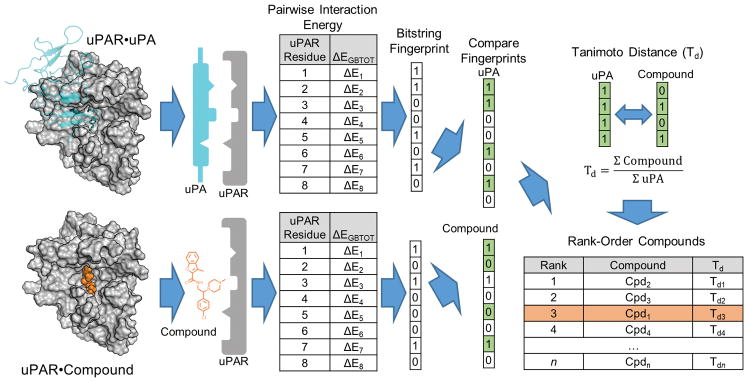
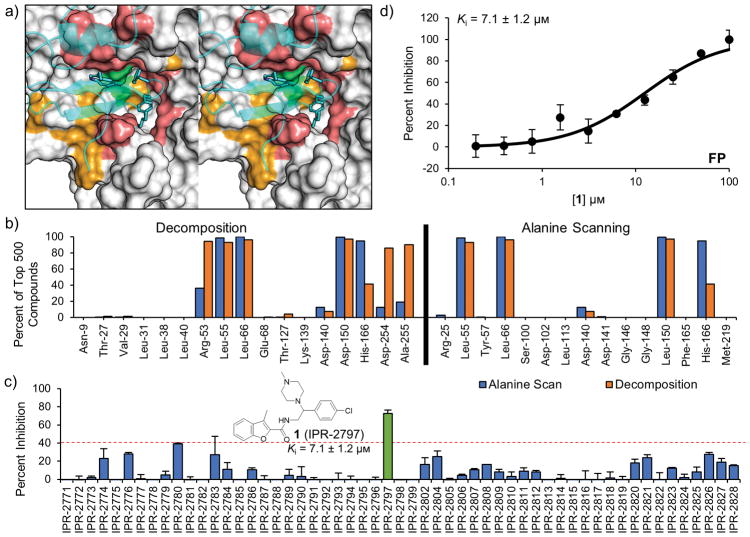
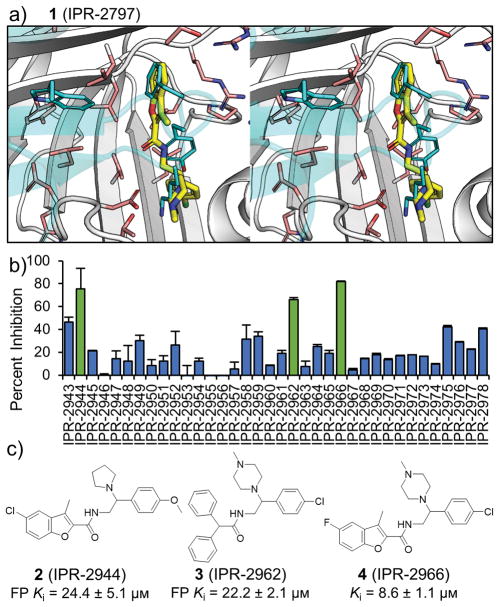
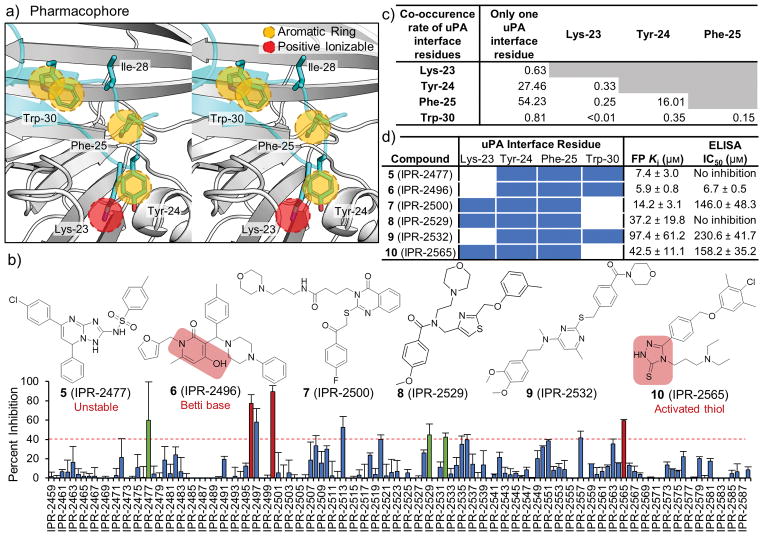
Tight protein-protein interactions (Kd <100 nm) that occur over a large binding interface (>1000 Å2 ) are highly challenging to disrupt with small molecules. Historically, the design of small molecules to inhibit protein-protein interactions has focused on mimicking the position of interface protein ligand side chains. Here, we explore mimicry of the pairwise intermolecular interactions of the native protein ligand with residues of the protein receptor to enrich commercial libraries for small-molecule inhibitors of tight protein-protein interactions. We use the high-affinity interaction (Kd =1 nm) between the urokinase receptor (uPAR) and its ligand urokinase (uPA) to test our methods. We introduce three methods for rank-ordering small molecules docked to uPAR: 1) a new fingerprint approach that represents uPA's pairwise interaction energies with uPAR residues; 2) a pharmacophore approach to identify small molecules that mimic the position of uPA interface residues; and 3) a combined fingerprint and pharmacophore approach. Our work led to small molecules with novel chemotypes that inhibited a tight uPAR⋅uPA protein-protein interaction with single-digit micromolar IC50 values. We also report the extensive work that identified several of the hits as either lacking stability, thiol reactive, or redox active. This work suggests that mimicking the binding profile of the native ligand and the position of interface residues can be an effective strategy to enrich commercial libraries for small-molecule inhibitors of tight protein-protein interactions.
Keywords: protein-protein interactions; small molecules; urokinase receptor; virtual screening.
© 2017 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.
Figures
Similar articles
-
Chemical Space Overlap with Critical Protein-Protein Interface Residues in Commercial and Specialized Small-Molecule Libraries.ChemMedChem. 2019 Jan 8;14(1):119-131. doi: 10.1002/cmdc.201800537. Epub 2018 Dec 20. ChemMedChem. 2019. PMID: 30548204 Free PMC article.
-
Small Molecules Engage Hot Spots through Cooperative Binding To Inhibit a Tight Protein-Protein Interaction.Biochemistry. 2017 Mar 28;56(12):1768-1784. doi: 10.1021/acs.biochem.6b01039. Epub 2017 Mar 17. Biochemistry. 2017. PMID: 28186725 Free PMC article.
-
A new class of orthosteric uPAR·uPA small-molecule antagonists are allosteric inhibitors of the uPAR·vitronectin interaction.ACS Chem Biol. 2015 Jun 19;10(6):1521-34. doi: 10.1021/cb500832q. Epub 2015 Mar 31. ACS Chem Biol. 2015. PMID: 25671694 Free PMC article.
-
Structure, function and expression on blood and bone marrow cells of the urokinase-type plasminogen activator receptor, uPAR.Stem Cells. 1997;15(6):398-408. doi: 10.1002/stem.150398. Stem Cells. 1997. PMID: 9402652 Review.
-
Urokinase plasminogen activator system as a potential target for cancer therapy.Future Oncol. 2009 Nov;5(9):1487-99. doi: 10.2217/fon.09.108. Future Oncol. 2009. PMID: 19903074 Review.
Cited by
-
Therapeutic potential of targeting the Eph/ephrin signaling complex.Int J Biochem Cell Biol. 2018 Dec;105:123-133. doi: 10.1016/j.biocel.2018.10.006. Epub 2018 Oct 19. Int J Biochem Cell Biol. 2018. PMID: 30343150 Free PMC article. Review.
-
Unconventional Secondary Structure Mimics: Ladder-Rungs.Angew Chem Int Ed Engl. 2020 Jun 8;59(24):9398-9402. doi: 10.1002/anie.202002639. Epub 2020 Apr 20. Angew Chem Int Ed Engl. 2020. PMID: 32176815 Free PMC article.
-
Targeting the Urokinase-Type Plasminogen Activator Receptor (uPAR) in Human Diseases With a View to Non-invasive Imaging and Therapeutic Intervention.Front Cell Dev Biol. 2021 Aug 20;9:732015. doi: 10.3389/fcell.2021.732015. eCollection 2021. Front Cell Dev Biol. 2021. PMID: 34490277 Free PMC article. Review.
-
Urokinase-type plasminogen activator receptor (uPAR) as a therapeutic target in cancer.J Transl Med. 2022 Mar 18;20(1):135. doi: 10.1186/s12967-022-03329-3. J Transl Med. 2022. PMID: 35303878 Free PMC article. Review.
-
Chemical Space Overlap with Critical Protein-Protein Interface Residues in Commercial and Specialized Small-Molecule Libraries.ChemMedChem. 2019 Jan 8;14(1):119-131. doi: 10.1002/cmdc.201800537. Epub 2018 Dec 20. ChemMedChem. 2019. PMID: 30548204 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
