

Compound heterozygous mutations in UBA5 causing early-onset epileptic encephalopathy in two sisters
- PMID: 28965491
- PMCID: PMC5623963
- DOI: 10.1186/s12881-017-0466-8
Compound heterozygous mutations in UBA5 causing early-onset epileptic encephalopathy in two sisters
Abstract
Background: Epileptic encephalopathies are a group of childhood epilepsies that display high phenotypic and genetic heterogeneity. The recent, extensive use of next-generation sequencing has identified a large number of genes in epileptic encephalopathies, including UBA5 in which biallelic mutations were first described as pathogenic in 2016 (Colin E et al., Am J Hum Genet 99(3):695-703, 2016. Muona M et al., Am J Hum Genet 99(3):683-694, 2016). UBA5 encodes an activating enzyme for a post-translational modification mechanism known as ufmylation, and is the first gene from the ufmylation pathway that is linked to disease.
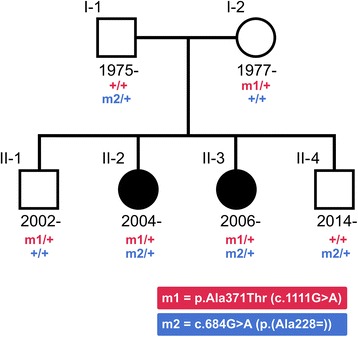
Case presentation: We sequenced the genomes of two sisters with early-onset epileptic encephalopathy along with their unaffected parents in an attempt to find a genetic cause for their condition. The sisters, born in 2004 and 2006, presented with infantile spasms at six months of age, which later progressed to recurrent, treatment-resistant seizures. We detected a compound heterozygous genotype in UBA5 in the sisters, a genotype not seen elsewhere in an Icelandic reference set of 30,067 individuals nor in public databases. One of the mutations, c.684G > A, is a paternally inherited exonic splicing mutation, occuring at the last nucleotide of exon 7 of UBA5. The mutation is predicted to disrupt the splice site, resulting in loss-of-function of one allele of UBA5. The second mutation is a maternally inherited missense mutation, p.Ala371Thr, previously reported as pathogenic when in compound heterozygosity with a loss-of-function mutation in UBA5 and is believed to produce a hypomorphic allele. Supportive of this, we have identified three adult Icelanders homozygous for the p.Ala371Thr mutation who show no signs of neurological disease.
Conclusions: We describe compound heterozygous mutations in the UBA5 gene in two sisters with early-onset epileptic encephalopathy. To our knowledge, this is the first description of mutations in UBA5 since the initial discovery that pathogenic biallelic variants in the gene cause early-onset epileptic encephalopathy. We further provide confirmatory evidence that p.Ala371Thr is a hypomorphic mutation, by presenting three adult homozygotes who show no signs of neurological disease.
Keywords: Case report; Epileptic encephalopathy; Exonic splicing mutation; Hypomorphic mutation; UBA5; Ufmylation.
Conflict of interest statement
Ethics approval and consent to participate
The study was approved by the National Bioethics Committee in Iceland. Written, informed consent to participate was obtained for all participants, or their guardians, before blood samples were drawn.
Consent for publication
All participants, or their guardians, provided written informed consent for publication of medical data, medical imaging and genetic data.
Competing interests
GAA, BOJ, GS, AO, RPK, SB, SAG, GM, GAT, JS, OTM, AJ, ASJ, AS, DFG, UT, PS and KS are full-time employees of deCODE Genetics/Amgen, Inc. SEM and RA declare no conflict of interest.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
Similar articles
-
A description of novel variants and review of phenotypic spectrum in UBA5-related early epileptic encephalopathy.Cold Spring Harb Mol Case Stud. 2021 Jun 11;7(3):a005827. doi: 10.1101/mcs.a005827. Print 2021 Jun. Cold Spring Harb Mol Case Stud. 2021. PMID: 33811063 Free PMC article.
-
Biallelic Variants in UBA5 Link Dysfunctional UFM1 Ubiquitin-like Modifier Pathway to Severe Infantile-Onset Encephalopathy.Am J Hum Genet. 2016 Sep 1;99(3):683-694. doi: 10.1016/j.ajhg.2016.06.020. Epub 2016 Aug 18. Am J Hum Genet. 2016. PMID: 27545674 Free PMC article.
-
Biallelic loss-of-function UBA5 mutations in a patient with intractable West syndrome and profound failure to thrive.Epileptic Disord. 2018 Aug 1;20(4):313-318. doi: 10.1684/epd.2018.0981. Epileptic Disord. 2018. PMID: 30078785
-
MED13 mutation: A novel cause of developmental and epileptic encephalopathy with infantile spasms.Seizure. 2022 Oct;101:211-217. doi: 10.1016/j.seizure.2022.09.002. Epub 2022 Sep 3. Seizure. 2022. PMID: 36087421 Review.
-
Severe early-onset developmental and epileptic encephalopathy (DEE) associated with novel compound heterozygous pathogenic variants in SLC25A22: Case report and literature review.Seizure. 2019 Aug;70:56-58. doi: 10.1016/j.seizure.2019.06.029. Epub 2019 Jun 27. Seizure. 2019. PMID: 31279168 Review. No abstract available.
Cited by
-
Transcriptomic analyses of human brains with Alzheimer's disease identified dysregulated epilepsy-causing genes.medRxiv [Preprint]. 2025 Jan 31:2025.01.02.25319900. doi: 10.1101/2025.01.02.25319900. medRxiv. 2025. Update in: Epilepsy Behav. 2025 Jul;168:110421. doi: 10.1016/j.yebeh.2025.110421. PMID: 39974070 Free PMC article. Updated. Preprint.
-
A homozygous loss-of-function mutation leading to CYBC1 deficiency causes chronic granulomatous disease.Nat Commun. 2018 Oct 25;9(1):4447. doi: 10.1038/s41467-018-06964-x. Nat Commun. 2018. PMID: 30361506 Free PMC article.
-
Next-Generation Sequencing Strategies.Cold Spring Harb Perspect Med. 2019 Jul 1;9(7):a025791. doi: 10.1101/cshperspect.a025791. Cold Spring Harb Perspect Med. 2019. PMID: 30323017 Free PMC article. Review.
-
UFMylation: A Potential Modification for Neurological Diseases.Curr Neuropharmacol. 2025;23(8):907-917. doi: 10.2174/011570159X340639240905092813. Curr Neuropharmacol. 2025. PMID: 39757643 Free PMC article. Review.
-
A description of novel variants and review of phenotypic spectrum in UBA5-related early epileptic encephalopathy.Cold Spring Harb Mol Case Stud. 2021 Jun 11;7(3):a005827. doi: 10.1101/mcs.a005827. Print 2021 Jun. Cold Spring Harb Mol Case Stud. 2021. PMID: 33811063 Free PMC article.
References
-
- Online Mendelian Inheritance in Man, OMIM®. McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University (Baltimore, MD). 1995–2017. https://www.omim.org/. Accessed May 2017.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
