

Myophosphorylase (PYGM) mutations determined by next generation sequencing in a cohort from Turkey with McArdle disease
- PMID: 28967462
- PMCID: PMC5698850
- DOI: 10.1016/j.nmd.2017.06.004
Myophosphorylase (PYGM) mutations determined by next generation sequencing in a cohort from Turkey with McArdle disease
Abstract
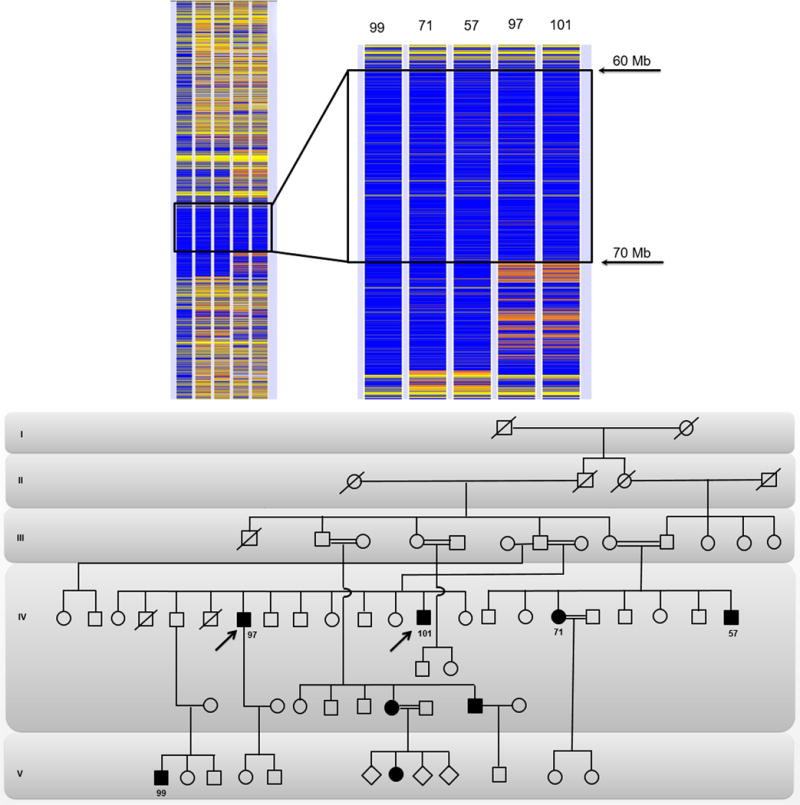
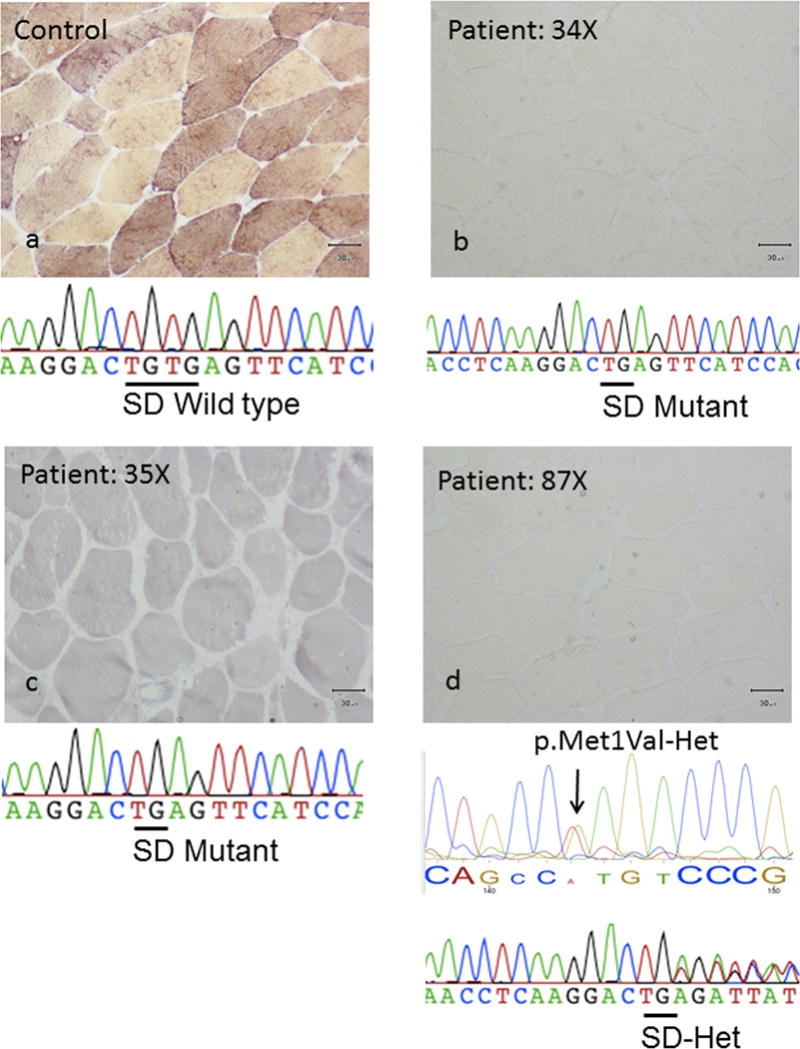
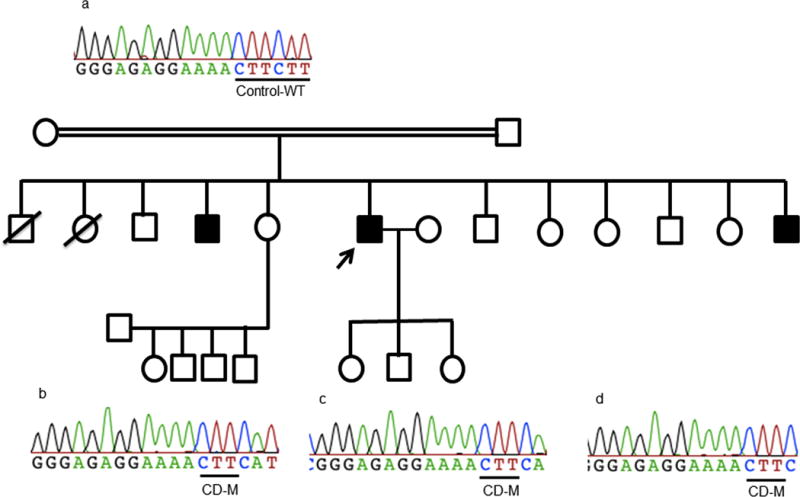
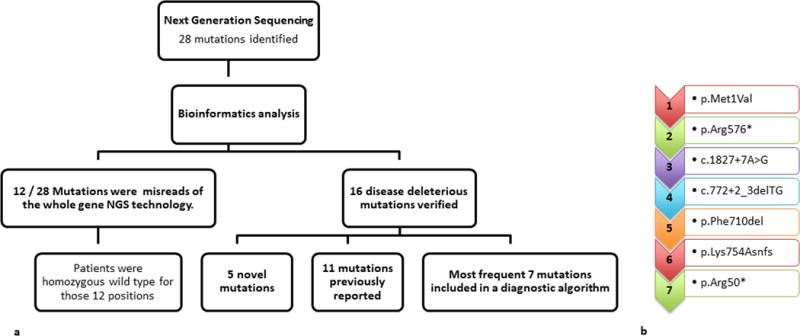
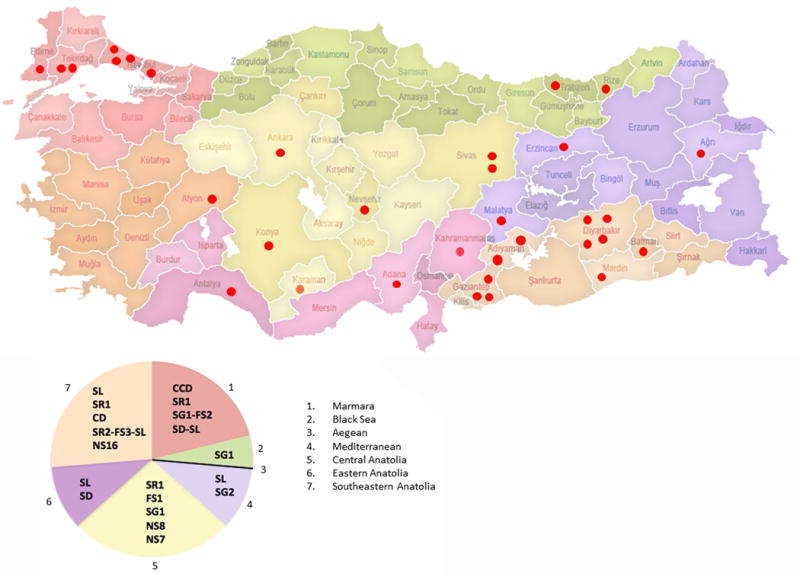
This study aimed to identify PYGM mutations in patients with McArdle disease from Turkey by next generation sequencing (NGS). Genomic DNA was extracted from the blood of the McArdle patients (n = 67) and unrelated healthy volunteers (n = 53). The PYGM gene was sequenced with NGS and the observed mutations were validated by direct Sanger sequencing. A diagnostic algorithm was developed for patients with suspected McArdle disease. A total of 16 deleterious PYGM mutations were identified, of which 5 were novel, including 1 splice-site donor, 1 frame-shift, and 3 non-synonymous variants. The p.Met1Val (27-patients/11-families) was the most common PYGM mutation, followed by p.Arg576* (6/4), c.1827+7A>G (5/4), c.772+2_3delTG (5/3), p.Phe710del (4/2), p.Lys754Asnfs (2/1), and p.Arg50* (1/1). A molecular diagnostic flowchart is proposed for the McArdle patients in Turkey, covering the 6 most common PYGM mutations found in Turkey as well as the most common mutation in Europe. The diagnostic algorithm may alleviate the need for muscle biopsies in 77.6% of future patients. A prevalence of any of the mutations to a geographical region in Turkey was not identified. Furthermore, the NGS approach to sequence the entire PYGM gene was successful in detecting a common missense mutation and discovering novel mutations in this population study.
Keywords: Genomics; Glycogenosis; Molecular screening; Novel mutation; Population specific; Rare muscle disorders.
Copyright © 2017 Elsevier B.V. All rights reserved.
Conflict of interest statement
Figures

References
-
- Darras BT, Friedman NR. Metabolic myopathies: a clinical approach; part II. Pediatr Neurol. 2000;22:171–81. - PubMed
-
- Chen YT. Glycogen storage diseases and other inherited disorders of carbohydrate metabolism. In: Kasper DL, Braunwald E, Fauci AS, Hauser SL, Longo DL, Jameson JL, editors. Harrison’s Principle of Internal Medicine. 16th. The McGraw-Hill Companies, Inc.; Pennsylvania, USA: 2005. pp. 2319–23.
-
- Lucia A, Nogales-Gadea G, Pérez M, Martín MA, Andreu AL, Arenas J. McArdle disease: what do neurologists need to know? Nat Clin Pract Neurol. 2008;4:568–77. - PubMed
-
- Deschauer M, Opalka JR, Lindner A, Zierz S. A novel nonsense mutation (R269X) in the myophosphorylase gene in a patient with McArdle disease. Mol Genet Metab. 2001;74:489–91. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
