

Assessment of variation in microbial community amplicon sequencing by the Microbiome Quality Control (MBQC) project consortium
- PMID: 28967885
- PMCID: PMC5839636
- DOI: 10.1038/nbt.3981
Assessment of variation in microbial community amplicon sequencing by the Microbiome Quality Control (MBQC) project consortium
Abstract
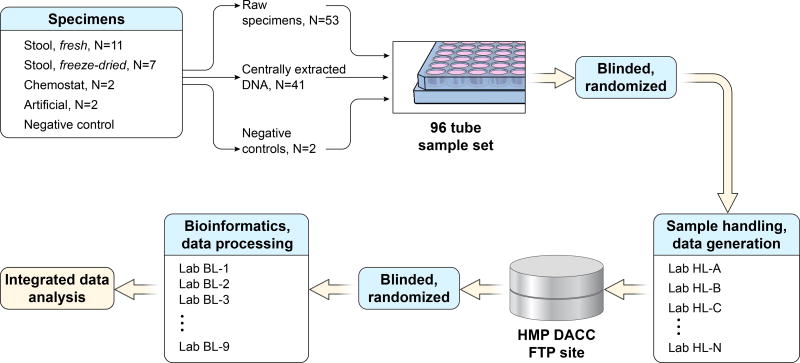
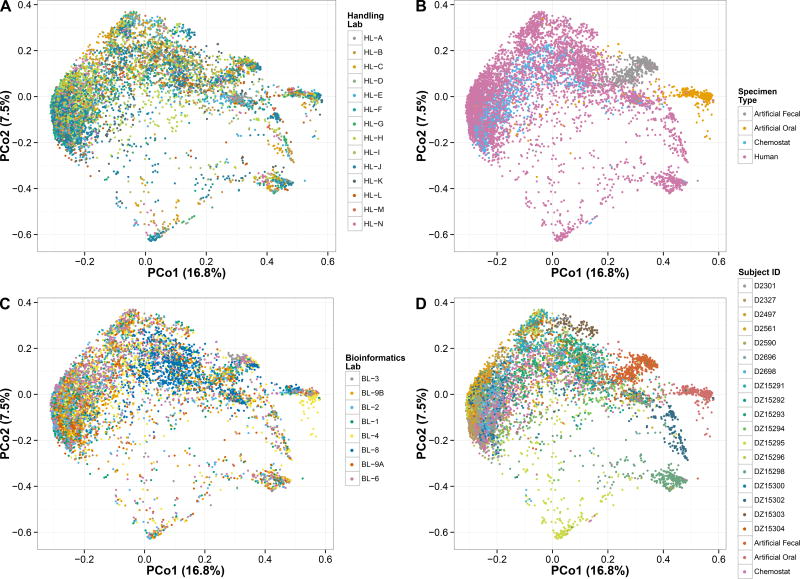
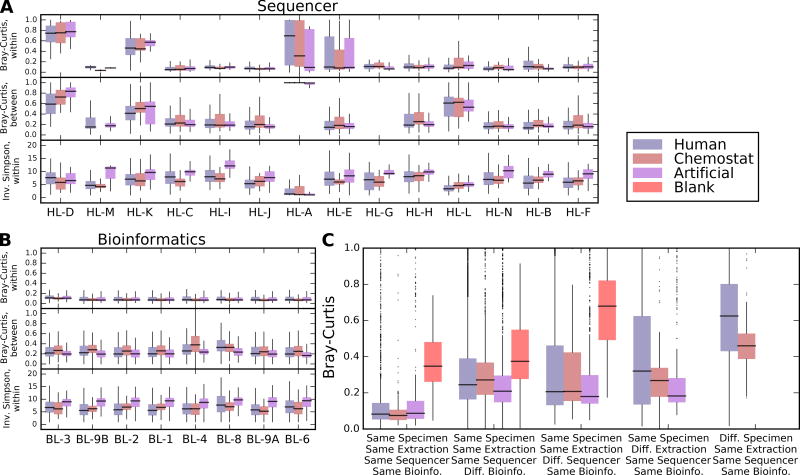
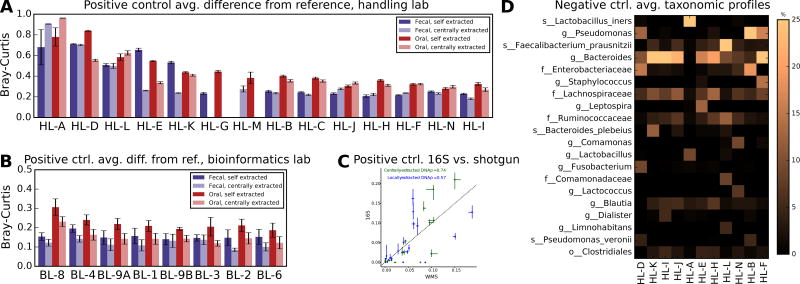
In order for human microbiome studies to translate into actionable outcomes for health, meta-analysis of reproducible data from population-scale cohorts is needed. Achieving sufficient reproducibility in microbiome research has proven challenging. We report a baseline investigation of variability in taxonomic profiling for the Microbiome Quality Control (MBQC) project baseline study (MBQC-base). Blinded specimen sets from human stool, chemostats, and artificial microbial communities were sequenced by 15 laboratories and analyzed using nine bioinformatics protocols. Variability depended most on biospecimen type and origin, followed by DNA extraction, sample handling environment, and bioinformatics. Analysis of artificial community specimens revealed differences in extraction efficiency and bioinformatic classification. These results may guide researchers in experimental design choices for gut microbiome studies.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
