Critical Assessment of Metagenome Interpretation-a benchmark of metagenomics software
- PMID: 28967888
- PMCID: PMC5903868
- DOI: 10.1038/nmeth.4458
Critical Assessment of Metagenome Interpretation-a benchmark of metagenomics software
Abstract
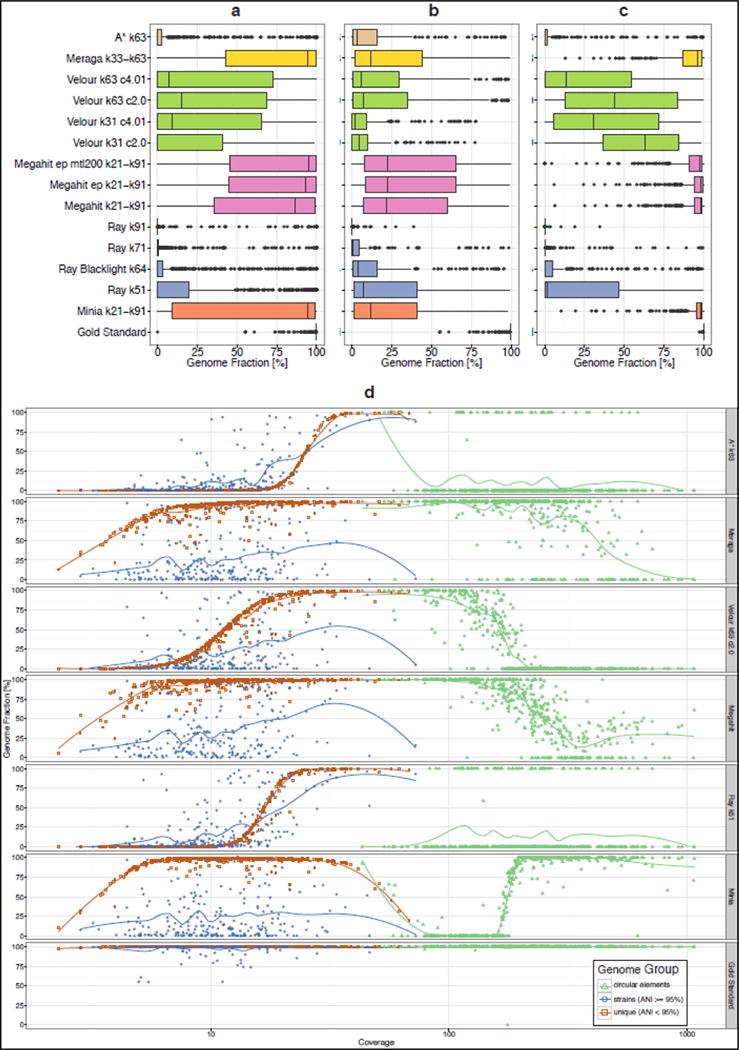
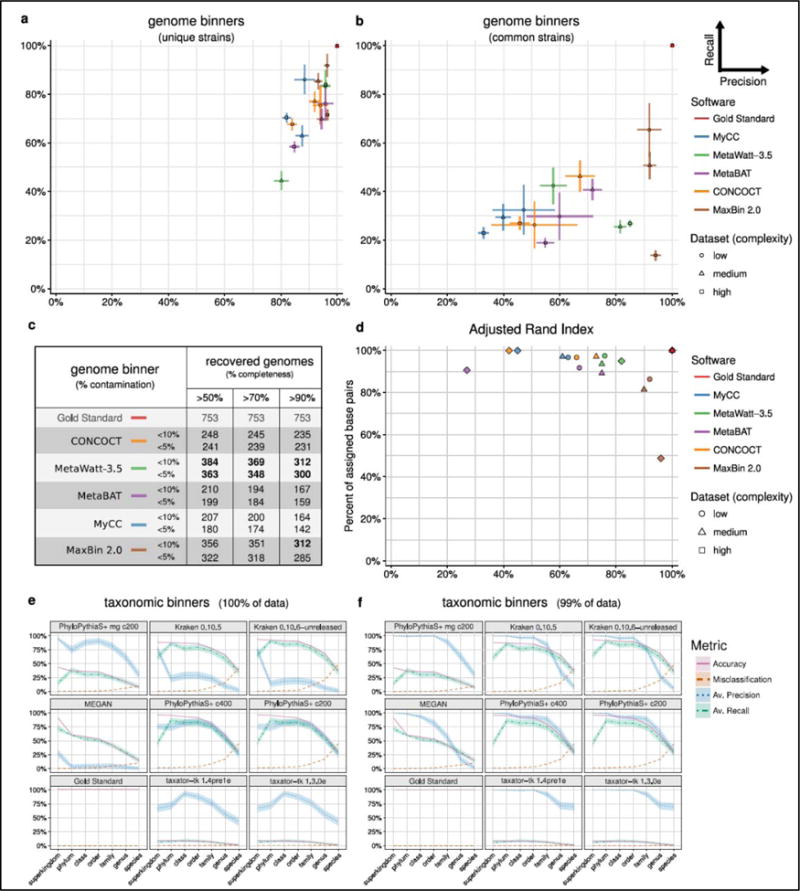
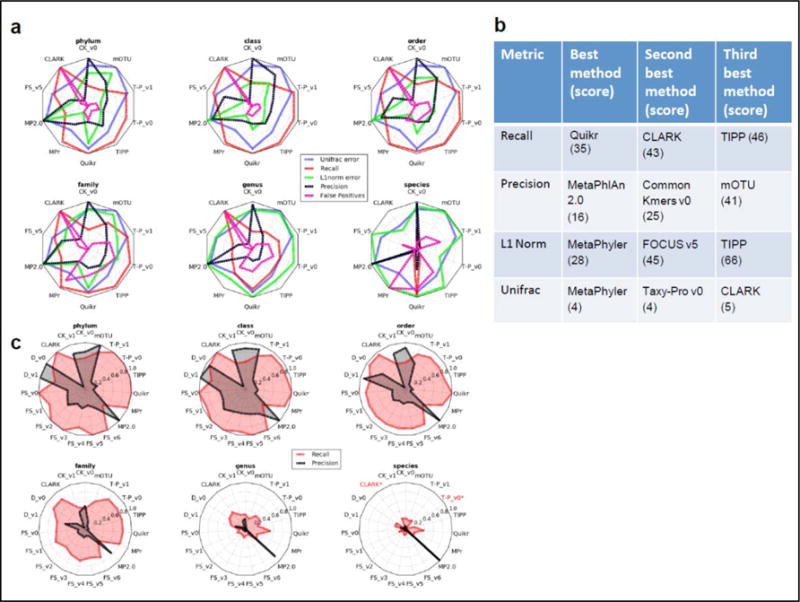
Methods for assembly, taxonomic profiling and binning are key to interpreting metagenome data, but a lack of consensus about benchmarking complicates performance assessment. The Critical Assessment of Metagenome Interpretation (CAMI) challenge has engaged the global developer community to benchmark their programs on highly complex and realistic data sets, generated from ∼700 newly sequenced microorganisms and ∼600 novel viruses and plasmids and representing common experimental setups. Assembly and genome binning programs performed well for species represented by individual genomes but were substantially affected by the presence of related strains. Taxonomic profiling and binning programs were proficient at high taxonomic ranks, with a notable performance decrease below family level. Parameter settings markedly affected performance, underscoring their importance for program reproducibility. The CAMI results highlight current challenges but also provide a roadmap for software selection to answer specific research questions.
Figures