

Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis
- PMID: 28967920
- PMCID: PMC5677540
- DOI: 10.1038/nm.4407
Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis
Abstract
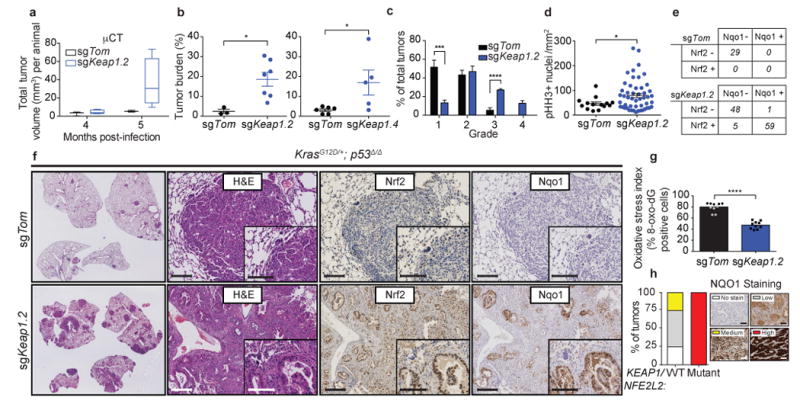
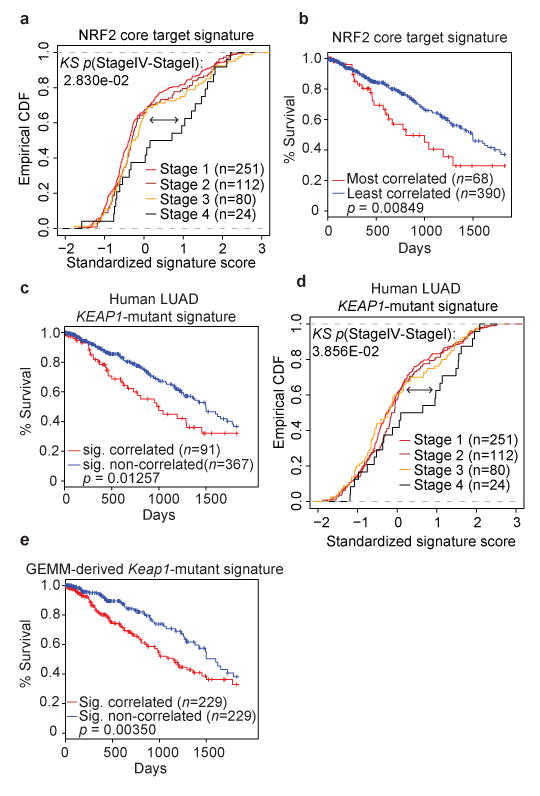
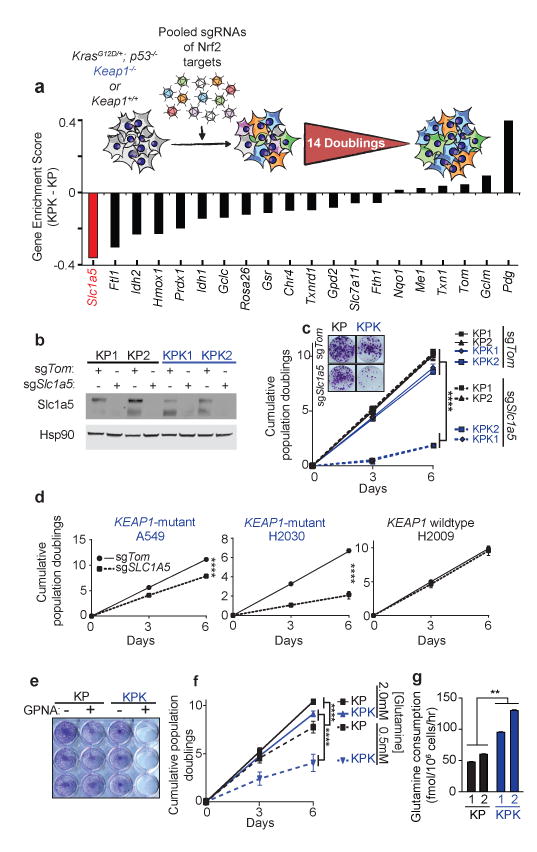
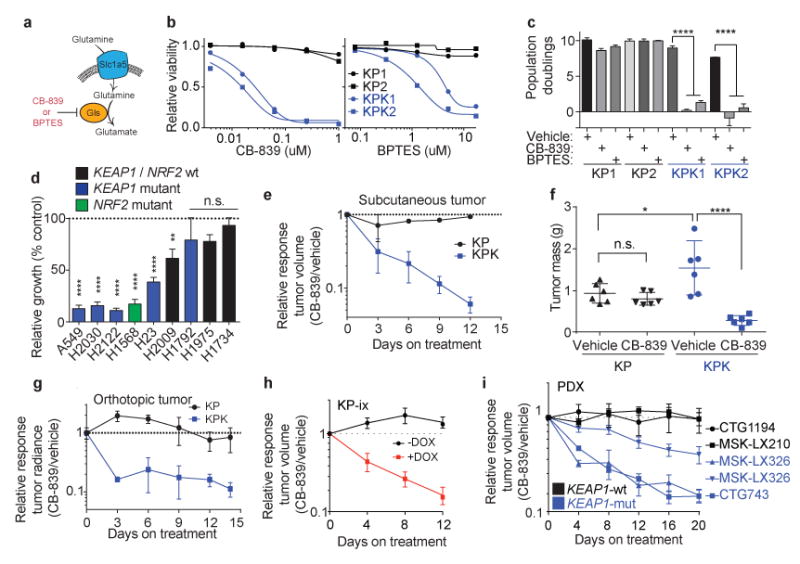
Treating KRAS-mutant lung adenocarcinoma (LUAD) remains a major challenge in cancer treatment given the difficulties associated with directly inhibiting the KRAS oncoprotein. One approach to addressing this challenge is to define mutations that frequently co-occur with those in KRAS, which themselves may lead to therapeutic vulnerabilities in tumors. Approximately 20% of KRAS-mutant LUAD tumors carry loss-of-function mutations in the KEAP1 gene encoding Kelch-like ECH-associated protein 1 (refs. 2, 3, 4), a negative regulator of nuclear factor erythroid 2-like 2 (NFE2L2; hereafter NRF2), which is the master transcriptional regulator of the endogenous antioxidant response. The high frequency of mutations in KEAP1 suggests an important role for the oxidative stress response in lung tumorigenesis. Using a CRISPR-Cas9-based approach in a mouse model of KRAS-driven LUAD, we examined the effects of Keap1 loss in lung cancer progression. We show that loss of Keap1 hyperactivates NRF2 and promotes KRAS-driven LUAD in mice. Through a combination of CRISPR-Cas9-based genetic screening and metabolomic analyses, we show that Keap1- or Nrf2-mutant cancers are dependent on increased glutaminolysis, and this property can be therapeutically exploited through the pharmacological inhibition of glutaminase. Finally, we provide a rationale for stratification of human patients with lung cancer harboring KRAS/KEAP1- or KRAS/NRF2-mutant lung tumors as likely to respond to glutaminase inhibition.
Figures
References
-
- Itoh K, et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236:313–322. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
