

Regulation of nitric oxide signaling by formation of a distal receptor-ligand complex
- PMID: 28967923
- PMCID: PMC5698159
- DOI: 10.1038/nchembio.2488
Regulation of nitric oxide signaling by formation of a distal receptor-ligand complex
Abstract
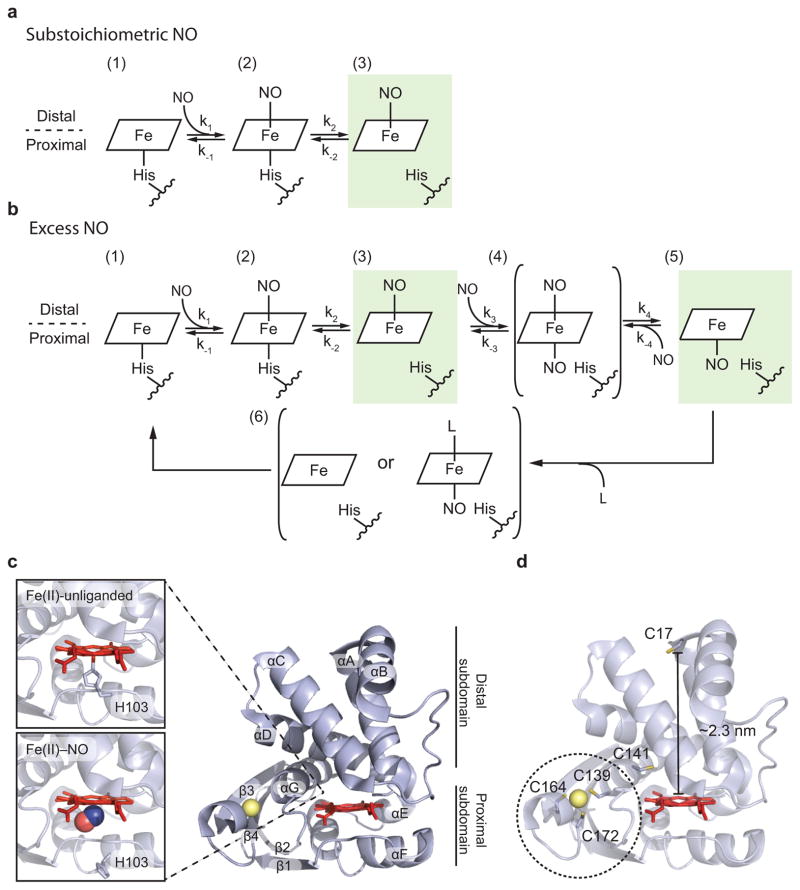
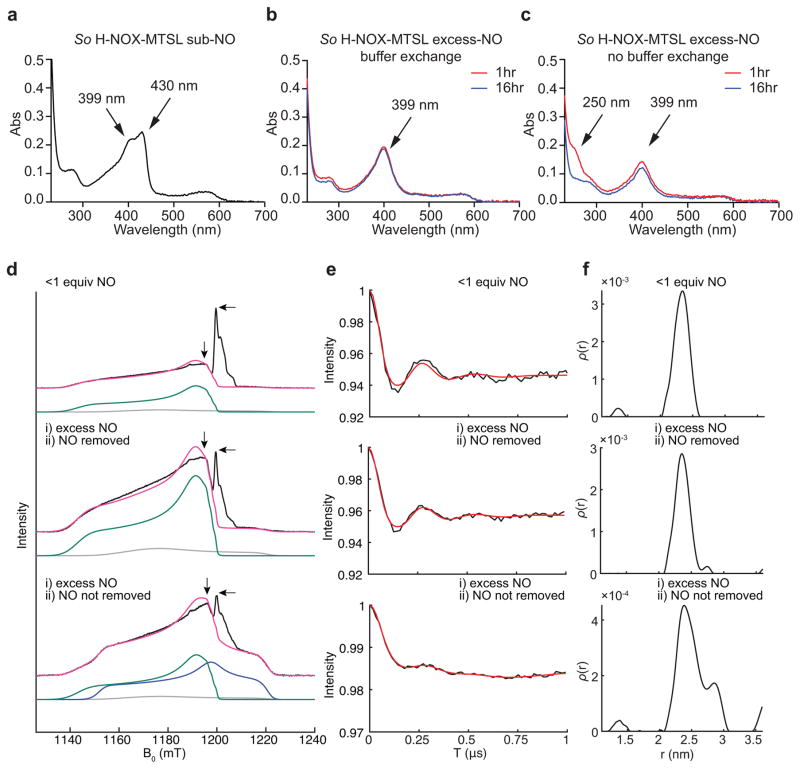
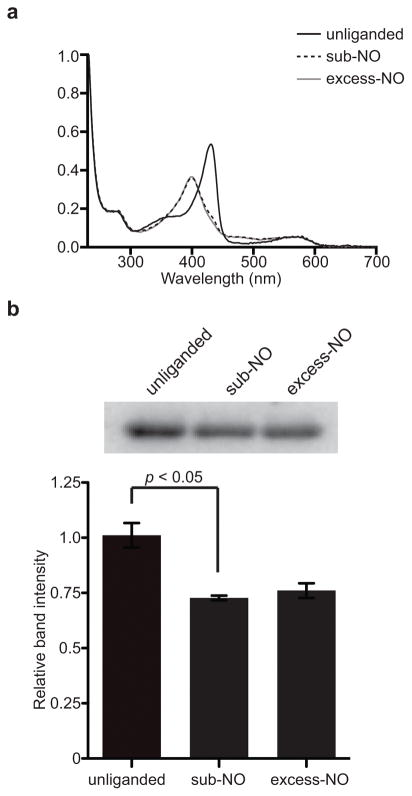
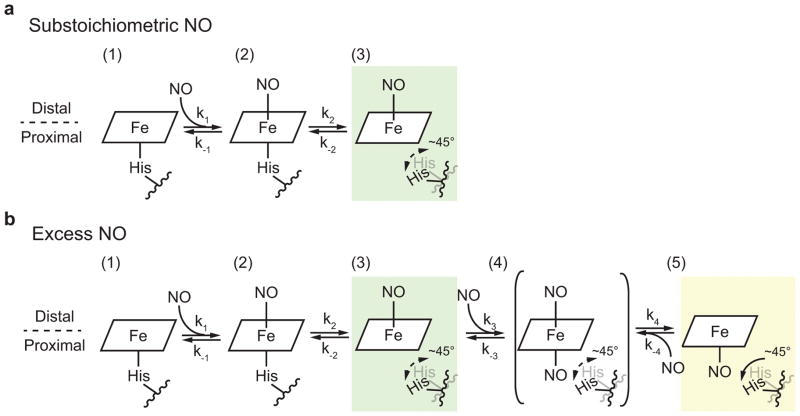
The binding of nitric oxide (NO) to the heme cofactor of heme-nitric oxide/oxygen binding (H-NOX) proteins can lead to the dissociation of the heme-ligating histidine residue and yield a five-coordinate nitrosyl complex, an important step for NO-dependent signaling. In the five-coordinate nitrosyl complex, NO can reside on either the distal or proximal side of the heme, which could have a profound influence over the lifetime of the in vivo signal. To investigate this central molecular question, we characterized the Shewanella oneidensis H-NOX (So H-NOX)-NO complex biophysically under limiting and excess NO conditions. The results show that So H-NOX preferably forms a distal NO species with both limiting and excess NO. Therefore, signal strength and complex lifetime in vivo will be dictated by the dissociation rate of NO from the distal complex and the rebinding of the histidine ligand to the heme.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
Similar articles
-
Structural insights into the role of iron-histidine bond cleavage in nitric oxide-induced activation of H-NOX gas sensor proteins.Proc Natl Acad Sci U S A. 2014 Oct 7;111(40):E4156-64. doi: 10.1073/pnas.1416936111. Epub 2014 Sep 24. Proc Natl Acad Sci U S A. 2014. PMID: 25253889 Free PMC article.
-
Nitric Oxide-Induced Conformational Changes Govern H-NOX and Histidine Kinase Interaction and Regulation in Shewanella oneidensis.Biochemistry. 2017 Mar 7;56(9):1274-1284. doi: 10.1021/acs.biochem.6b01133. Epub 2017 Feb 21. Biochemistry. 2017. PMID: 28170222
-
Is histidine dissociation a critical component of the NO/H-NOX signaling mechanism? Insights from X-ray absorption spectroscopy.Dalton Trans. 2012 Jul 14;41(26):7984-93. doi: 10.1039/c2dt30147d. Epub 2012 Mar 20. Dalton Trans. 2012. PMID: 22430114 Free PMC article.
-
Structural Insight into H-NOX Gas Sensing and Cognate Signaling Protein Regulation.Chembiochem. 2019 Jan 2;20(1):7-19. doi: 10.1002/cbic.201800478. Epub 2018 Nov 8. Chembiochem. 2019. PMID: 30320963 Review.
-
Nitric oxide-sensing H-NOX proteins govern bacterial communal behavior.Trends Biochem Sci. 2013 Nov;38(11):566-75. doi: 10.1016/j.tibs.2013.08.008. Epub 2013 Oct 7. Trends Biochem Sci. 2013. PMID: 24113192 Free PMC article. Review.
Cited by
-
Allosteric activation of the nitric oxide receptor soluble guanylate cyclase mapped by cryo-electron microscopy.Elife. 2019 Sep 30;8:e50634. doi: 10.7554/eLife.50634. Elife. 2019. PMID: 31566566 Free PMC article.
-
A new paradigm for gaseous ligand selectivity of hemoproteins highlighted by soluble guanylate cyclase.J Inorg Biochem. 2021 Jan;214:111267. doi: 10.1016/j.jinorgbio.2020.111267. Epub 2020 Oct 16. J Inorg Biochem. 2021. PMID: 33099233 Free PMC article. Review.
-
The Mechanism of Biochemical NO-Sensing: Insights from Computational Chemistry.Chemistry. 2022 Sep 1;28(49):e202200930. doi: 10.1002/chem.202200930. Epub 2022 Jul 11. Chemistry. 2022. PMID: 35670519 Free PMC article.
-
Nitric Oxide Binding Geometry in Heme-Proteins: Relevance for Signal Transduction.Antioxidants (Basel). 2024 May 29;13(6):666. doi: 10.3390/antiox13060666. Antioxidants (Basel). 2024. PMID: 38929104 Free PMC article. Review.
-
Structure/function of the soluble guanylyl cyclase catalytic domain.Nitric Oxide. 2018 Jul 1;77:53-64. doi: 10.1016/j.niox.2018.04.008. Epub 2018 Apr 25. Nitric Oxide. 2018. PMID: 29702251 Free PMC article. Review.
References
-
- Derbyshire ER, Marletta MA. Structure and regulation of soluble guanylate cyclase. Annu Rev Biochem. 2012;81:533–59. - PubMed
-
- Marletta MA. Nitric oxide synthase: function and mechanism. Adv Exp Med Biol. 1993;338:281–4. - PubMed
-
- Marletta MA, Hurshman AR, Rusche KM. Catalysis by nitric oxide synthase. Curr Opin Chem Biol. 1998;2:656–63. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
