

Subarachnoid blood acutely induces spreading depolarizations and early cortical infarction
- PMID: 28969382
- PMCID: PMC5841026
- DOI: 10.1093/brain/awx214
Subarachnoid blood acutely induces spreading depolarizations and early cortical infarction
Abstract
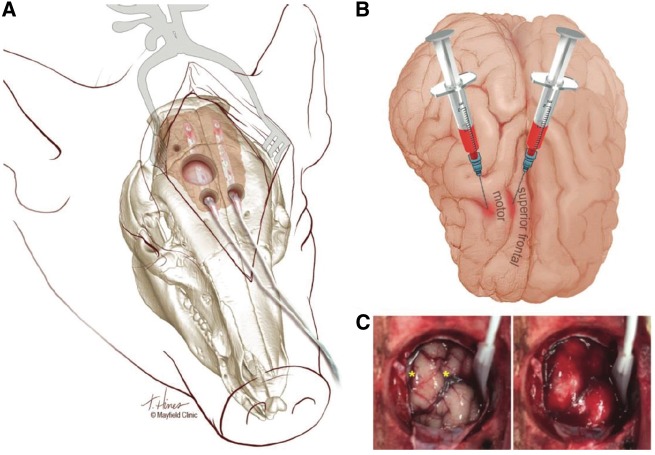
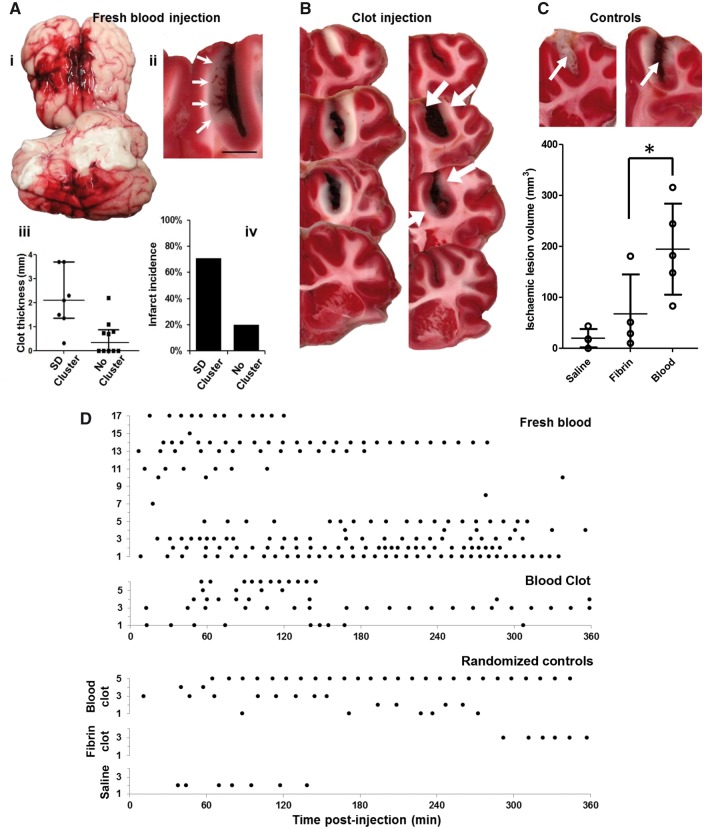
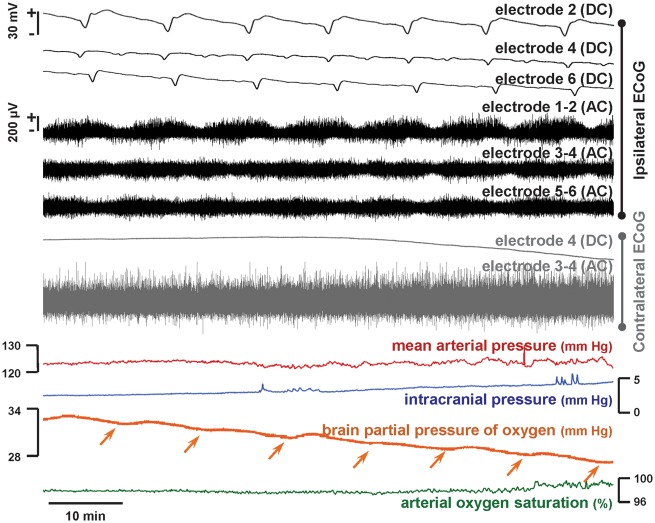
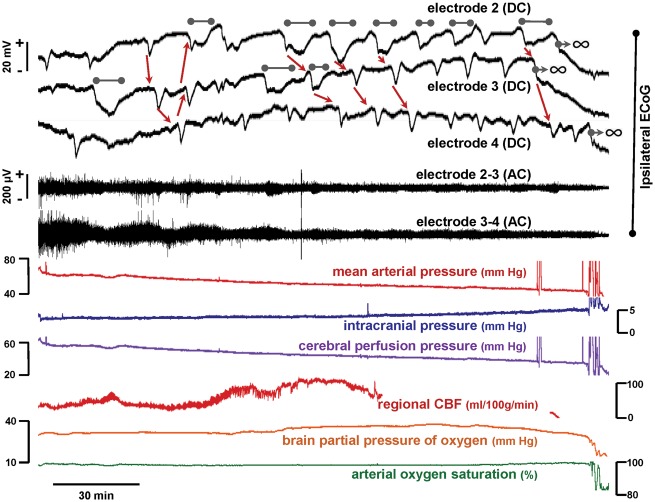
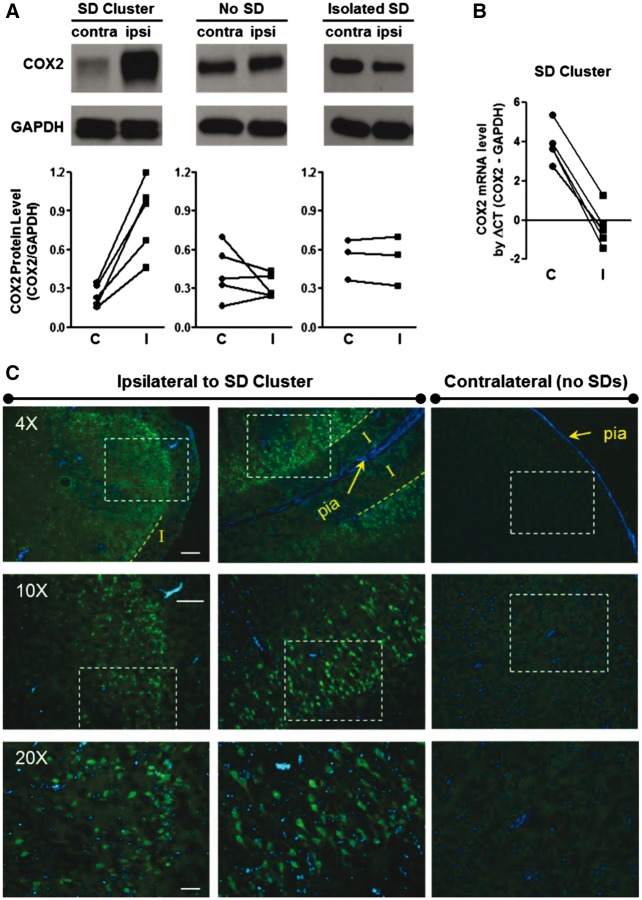
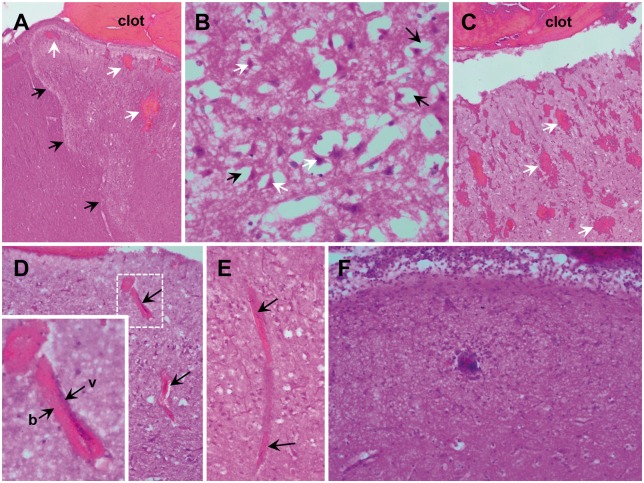
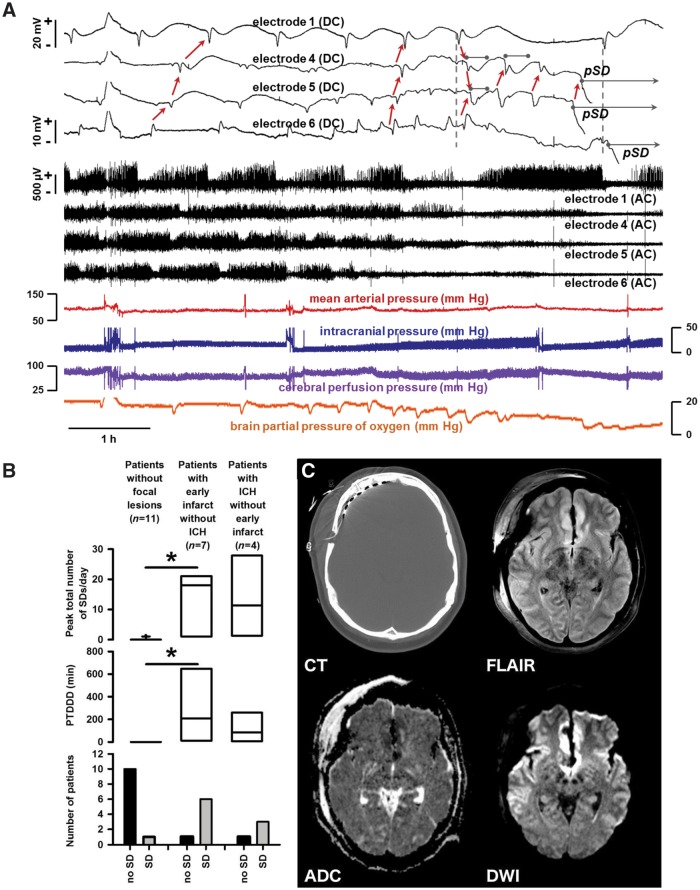
See Ghoshal and Claassen (doi:10.1093/brain/awx226) for a scientific commentary on this article. Early cortical infarcts are common in poor-grade patients after aneurysmal subarachnoid haemorrhage. There are no animal models of these lesions and mechanisms are unknown, although mass cortical spreading depolarizations are hypothesized as a requisite mechanism and clinical marker of infarct development. Here we studied acute sequelae of subarachnoid haemorrhage in the gyrencephalic brain of propofol-anaesthetized juvenile swine using subdural electrode strips (electrocorticography) and intraparenchymal neuromonitoring probes. Subarachnoid infusion of 1–2 ml of fresh blood at 200 µl/min over cortical sulci caused clusters of spreading depolarizations (count range: 12–34) in 7/17 animals in the ipsilateral but not contralateral hemisphere in 6 h of monitoring, without meaningful changes in other variables. Spreading depolarization clusters were associated with formation of sulcal clots (P < 0.01), a high likelihood of adjacent cortical infarcts (5/7 versus 2/10, P < 0.06), and upregulation of cyclooxygenase-2 in ipsilateral cortex remote from clots/infarcts. In a second cohort, infusion of 1 ml of clotted blood into a sulcus caused spreading depolarizations in 5/6 animals (count range: 4–20 in 6 h) and persistent thick clots with patchy or extensive infarction of circumscribed cortex in all animals. Infarcts were significantly larger after blood clot infusion compared to mass effect controls using fibrin clots of equal volume. Haematoxylin and eosin staining of infarcts showed well demarcated zones of oedema and hypoxic-ischaemic neuronal injury, consistent with acute infarction. The association of spreading depolarizations with early brain injury was then investigated in 23 patients [14 female; age (median, quartiles): 57 years (47, 63)] after repair of ruptured anterior communicating artery aneurysms by clip ligation (n = 14) or coiling (n = 9). Frontal electrocorticography [duration: 54 h (34, 66)] from subdural electrode strips was analysed over Days 0–3 after initial haemorrhage and magnetic resonance imaging studies were performed at ∼ 24–48 h after aneurysm treatment. Patients with frontal infarcts only and those with frontal infarcts and/or intracerebral haemorrhage were both significantly more likely to have spreading depolarizations (6/7 and 10/12, respectively) than those without frontal brain lesions (1/11, P’s < 0.05). These results suggest that subarachnoid clots in sulci/fissures are sufficient to induce spreading depolarizations and acute infarction in adjacent cortex. We hypothesize that the cellular toxicity and vasoconstrictive effects of depolarizations act in synergy with direct ischaemic effects of haemorrhage as mechanisms of infarct development. Results further validate spreading depolarizations as a clinical marker of early brain injury and establish a clinically relevant model to investigate causal pathologic sequences and potential therapeutic interventions.
Keywords: aneurysmal subarachnoid haemorrhage; brain infarction; cortical spreading depression; electroencephalography; intensive care.
Figures
Comment in
-
Spreading depolarization and acute ischaemia in subarachnoid haemorrhage: the role of mass depolarization waves.Brain. 2017 Oct 1;140(10):2527-2529. doi: 10.1093/brain/awx226. Brain. 2017. PMID: 28969392 No abstract available.
Similar articles
-
Similarities in the Electrographic Patterns of Delayed Cerebral Infarction and Brain Death After Aneurysmal and Traumatic Subarachnoid Hemorrhage.Transl Stroke Res. 2025 Feb;16(1):147-168. doi: 10.1007/s12975-024-01237-w. Epub 2024 Feb 23. Transl Stroke Res. 2025. PMID: 38396252 Free PMC article. Review.
-
Delayed ischaemic neurological deficits after subarachnoid haemorrhage are associated with clusters of spreading depolarizations.Brain. 2006 Dec;129(Pt 12):3224-37. doi: 10.1093/brain/awl297. Epub 2006 Oct 25. Brain. 2006. PMID: 17067993
-
Correlates of spreading depolarization in human scalp electroencephalography.Brain. 2012 Mar;135(Pt 3):853-68. doi: 10.1093/brain/aws010. Brain. 2012. PMID: 22366798 Free PMC article.
-
The negative ultraslow potential, electrophysiological correlate of infarction in the human cortex.Brain. 2018 Jun 1;141(6):1734-1752. doi: 10.1093/brain/awy102. Brain. 2018. PMID: 29668855 Free PMC article.
-
Impaired neurovascular coupling to ictal epileptic activity and spreading depolarization in a patient with subarachnoid hemorrhage: possible link to blood-brain barrier dysfunction.Epilepsia. 2012 Nov;53 Suppl 6(0 6):22-30. doi: 10.1111/j.1528-1167.2012.03699.x. Epilepsia. 2012. PMID: 23134492 Free PMC article. Review.
Cited by
-
Relevance of Porcine Stroke Models to Bridge the Gap from Pre-Clinical Findings to Clinical Implementation.Int J Mol Sci. 2020 Sep 8;21(18):6568. doi: 10.3390/ijms21186568. Int J Mol Sci. 2020. PMID: 32911769 Free PMC article. Review.
-
Small Vessels Are a Big Problem in Neurodegeneration and Neuroprotection.Front Neurol. 2019 Aug 16;10:889. doi: 10.3389/fneur.2019.00889. eCollection 2019. Front Neurol. 2019. PMID: 31474933 Free PMC article. Review.
-
Vasospasm-Induced Spreading Depolarization and/or Spreading-Depolarization-Induced Vasospasm After Subarachnoid Hemorrhage.Neurocrit Care. 2022 Jun;37(Suppl 1):5-7. doi: 10.1007/s12028-021-01373-3. Epub 2021 Oct 26. Neurocrit Care. 2022. PMID: 34704217 Free PMC article. No abstract available.
-
Brainstem and Cortical Spreading Depolarization in a Closed Head Injury Rat Model.Int J Mol Sci. 2021 Oct 28;22(21):11642. doi: 10.3390/ijms222111642. Int J Mol Sci. 2021. PMID: 34769073 Free PMC article.
-
Similarities in the Electrographic Patterns of Delayed Cerebral Infarction and Brain Death After Aneurysmal and Traumatic Subarachnoid Hemorrhage.Transl Stroke Res. 2025 Feb;16(1):147-168. doi: 10.1007/s12975-024-01237-w. Epub 2024 Feb 23. Transl Stroke Res. 2025. PMID: 38396252 Free PMC article. Review.
References
-
- Bretz JS, von Dincklage F, Woitzik J, Winkler MK, Major S, Dreier JP, et al.The Hijdra scale has a significant prognostic value for the functional outcome of Fisher grade 3 patients with subarachnoid hemorrhage. Clin Neuroradiol 2016. [Epub ahead of print]. doi: 10.1007/s00062-016-0509-0. - PubMed
-
- Broderick JP, Brott TG, Duldner JE, Tomsick T, Leach A. Initial and recurrent bleeding are the major causes of death following subarachnoid hemorrhage. Stroke 1994; 25: 1342–7. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
