

Mutations of AKT3 are associated with a wide spectrum of developmental disorders including extreme megalencephaly
- PMID: 28969385
- PMCID: PMC6080423
- DOI: 10.1093/brain/awx203
Mutations of AKT3 are associated with a wide spectrum of developmental disorders including extreme megalencephaly
Abstract
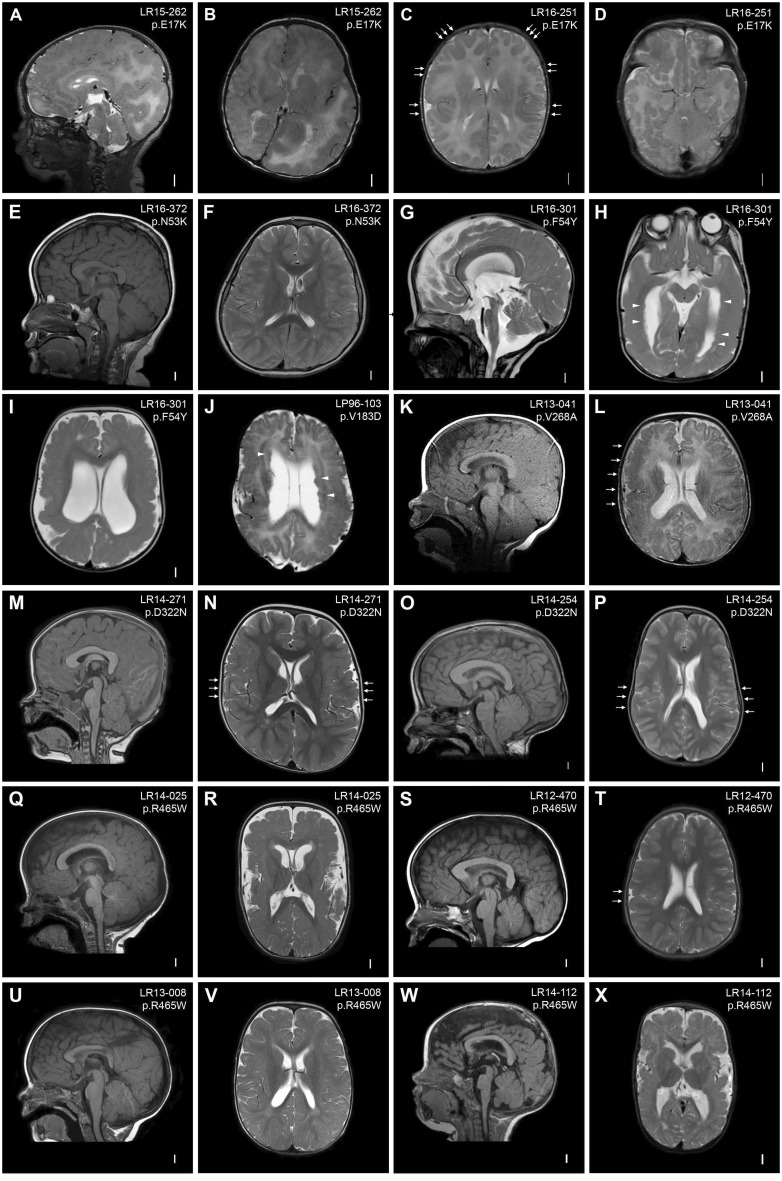
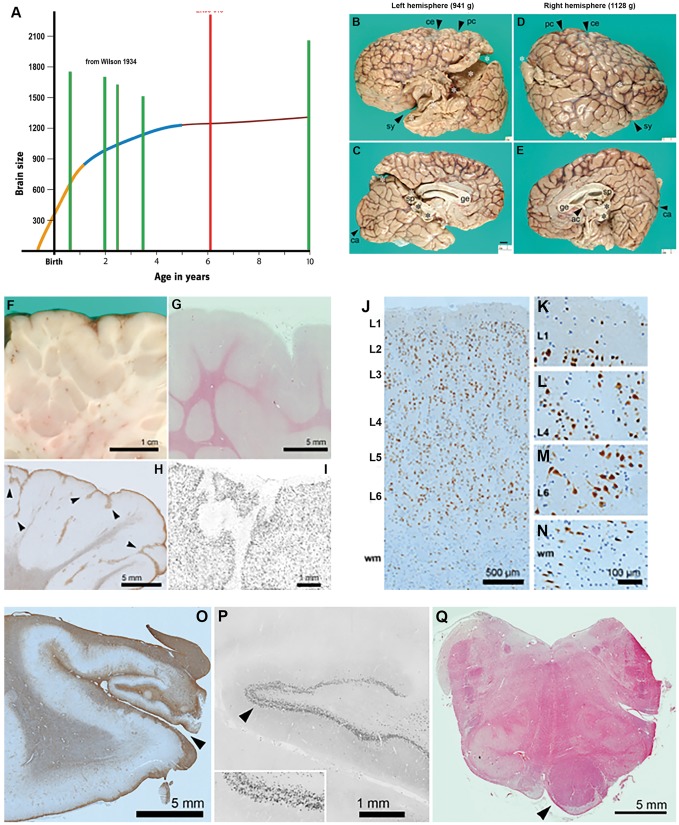
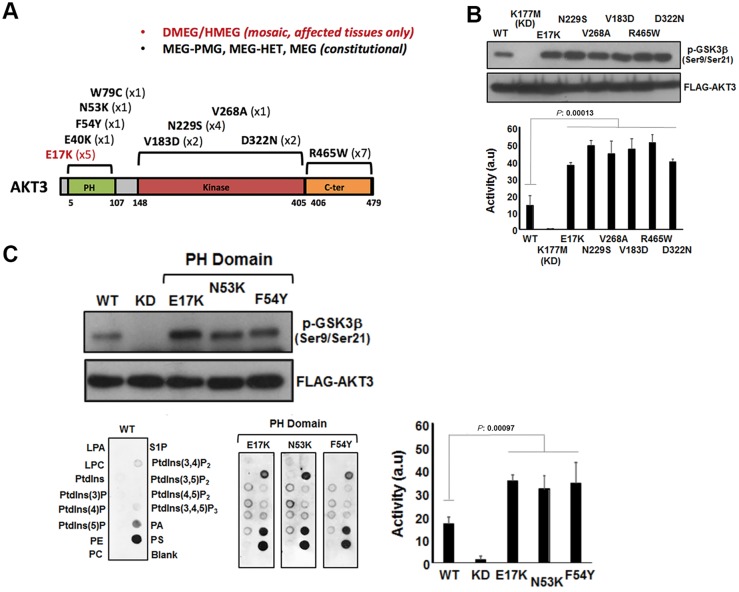
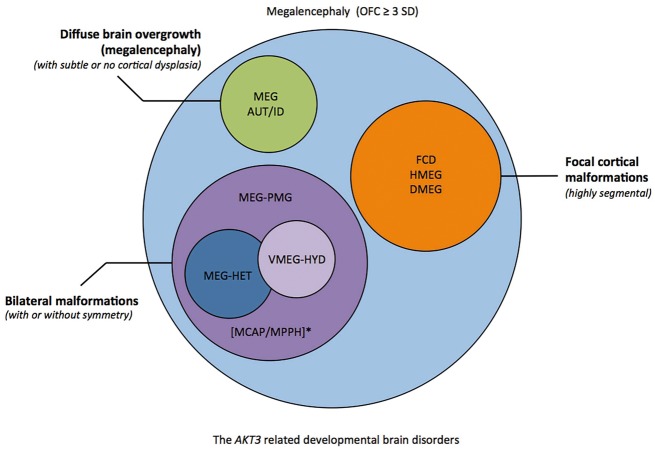
Mutations of genes within the phosphatidylinositol-3-kinase (PI3K)-AKT-MTOR pathway are well known causes of brain overgrowth (megalencephaly) as well as segmental cortical dysplasia (such as hemimegalencephaly, focal cortical dysplasia and polymicrogyria). Mutations of the AKT3 gene have been reported in a few individuals with brain malformations, to date. Therefore, our understanding regarding the clinical and molecular spectrum associated with mutations of this critical gene is limited, with no clear genotype-phenotype correlations. We sought to further delineate this spectrum, study levels of mosaicism and identify genotype-phenotype correlations of AKT3-related disorders. We performed targeted sequencing of AKT3 on individuals with these phenotypes by molecular inversion probes and/or Sanger sequencing to determine the type and level of mosaicism of mutations. We analysed all clinical and brain imaging data of mutation-positive individuals including neuropathological analysis in one instance. We performed ex vivo kinase assays on AKT3 engineered with the patient mutations and examined the phospholipid binding profile of pleckstrin homology domain localizing mutations. We identified 14 new individuals with AKT3 mutations with several phenotypes dependent on the type of mutation and level of mosaicism. Our comprehensive clinical characterization, and review of all previously published patients, broadly segregates individuals with AKT3 mutations into two groups: patients with highly asymmetric cortical dysplasia caused by the common p.E17K mutation, and patients with constitutional AKT3 mutations exhibiting more variable phenotypes including bilateral cortical malformations, polymicrogyria, periventricular nodular heterotopia and diffuse megalencephaly without cortical dysplasia. All mutations increased kinase activity, and pleckstrin homology domain mutants exhibited enhanced phospholipid binding. Overall, our study shows that activating mutations of the critical AKT3 gene are associated with a wide spectrum of brain involvement ranging from focal or segmental brain malformations (such as hemimegalencephaly and polymicrogyria) predominantly due to mosaic AKT3 mutations, to diffuse bilateral cortical malformations, megalencephaly and heterotopia due to constitutional AKT3 mutations. We also provide the first detailed neuropathological examination of a child with extreme megalencephaly due to a constitutional AKT3 mutation. This child has one of the largest documented paediatric brain sizes, to our knowledge. Finally, our data show that constitutional AKT3 mutations are associated with megalencephaly, with or without autism, similar to PTEN-related disorders. Recognition of this broad clinical and molecular spectrum of AKT3 mutations is important for providing early diagnosis and appropriate management of affected individuals, and will facilitate targeted design of future human clinical trials using PI3K-AKT pathway inhibitors.
Keywords: AKT3; epilepsy; hemimegalencephaly; megalencephaly; polymicrogyria.
© The Author (2017). Published by Oxford University Press on behalf of the Guarantors of Brain. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures
References
-
- Arya VB, Flanagan SE, Schober E, Rami-Merhar B, Ellard S, Hussain K. Activating AKT2 mutation: hypoinsulinemic hypoketotic hypoglycemia. J Clin Endocrinol Metab 2014; 99: 391–4. - PubMed
-
- Carpten JD, Faber AL, Horn C, Donoho GP, Briggs SL, Robbins CM, et al.A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature 2007; 448: 439–44. - PubMed
-
- Cho H, Mu J, Kim JK, Thorvaldsen JL, Chu Q, Crenshaw EB 3rd, et al.Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta). Science 2001; 292: 1728–31. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
