

Mitochondrial Respiration Is Reduced in Atherosclerosis, Promoting Necrotic Core Formation and Reducing Relative Fibrous Cap Thickness
- PMID: 28970293
- PMCID: PMC5701734
- DOI: 10.1161/ATVBAHA.117.310042
Mitochondrial Respiration Is Reduced in Atherosclerosis, Promoting Necrotic Core Formation and Reducing Relative Fibrous Cap Thickness
Erratum in
-
Correction to: Mitochondrial Respiration Is Reduced in Atherosclerosis, Promoting Necrotic Core Formation and Reducing Relative Fibrous Cap Thickness.Arterioscler Thromb Vasc Biol. 2018 Jul;38(7):e135. doi: 10.1161/ATV.0000000000000071. Arterioscler Thromb Vasc Biol. 2018. PMID: 29950387 Free PMC article. No abstract available.
Abstract
Objective: Mitochondrial DNA (mtDNA) damage is present in murine and human atherosclerotic plaques. However, whether endogenous levels of mtDNA damage are sufficient to cause mitochondrial dysfunction and whether decreasing mtDNA damage and improving mitochondrial respiration affects plaque burden or composition are unclear. We examined mitochondrial respiration in human atherosclerotic plaques and whether augmenting mitochondrial respiration affects atherogenesis.
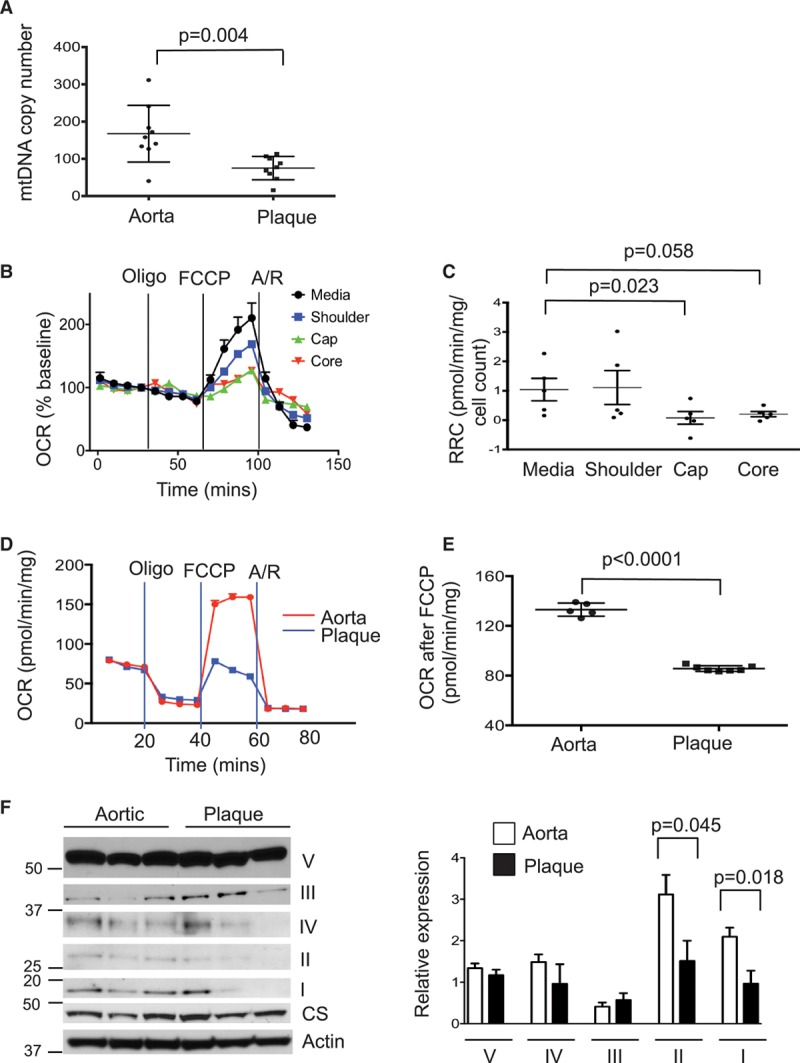
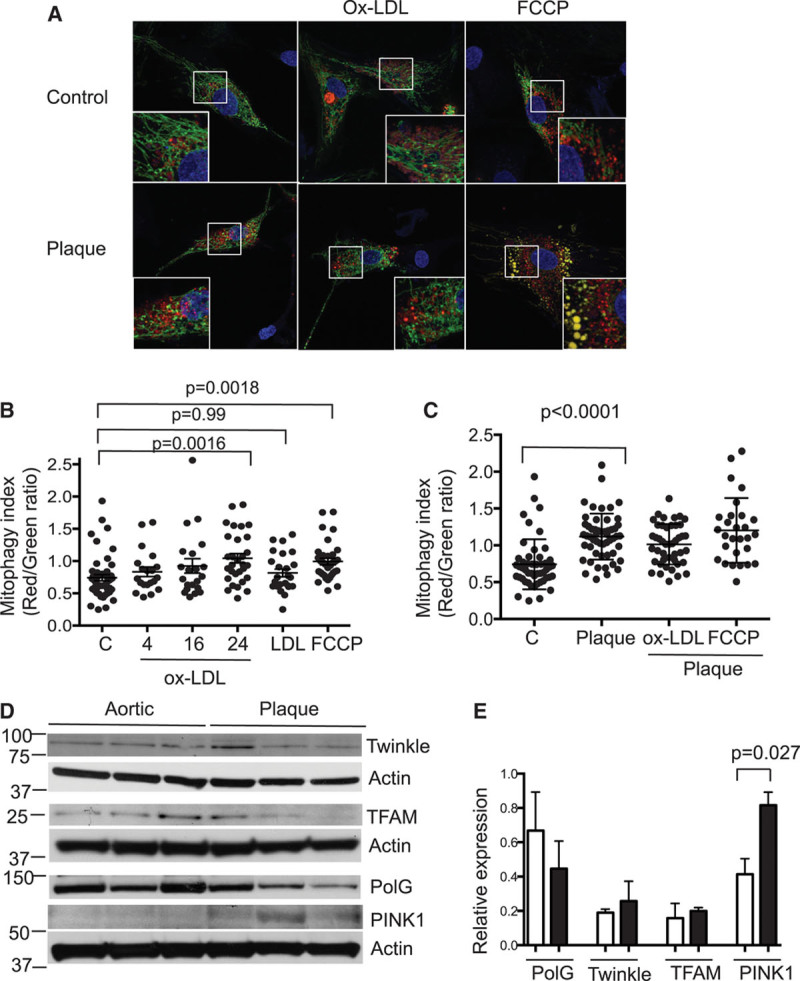
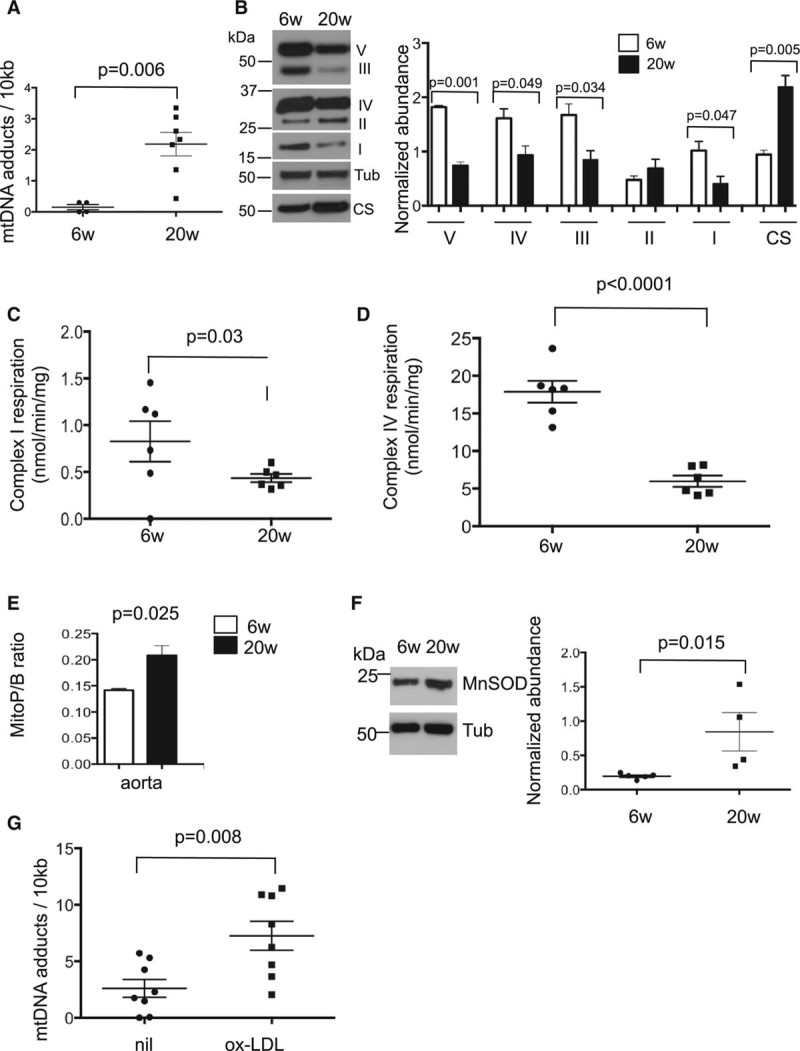
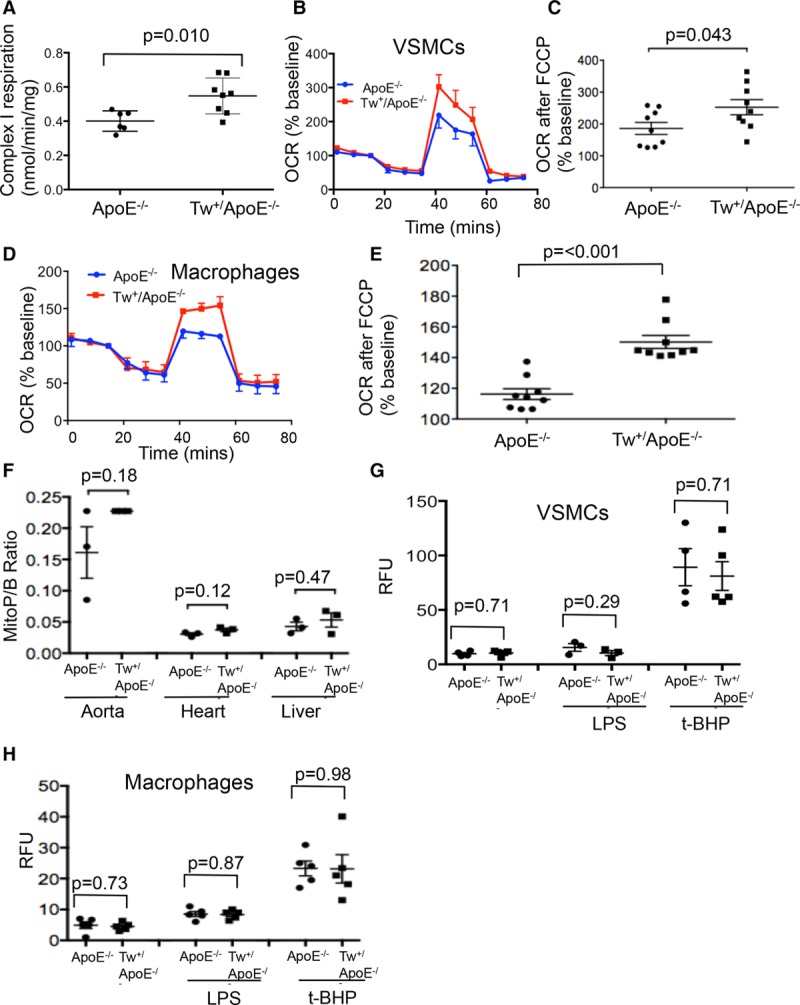
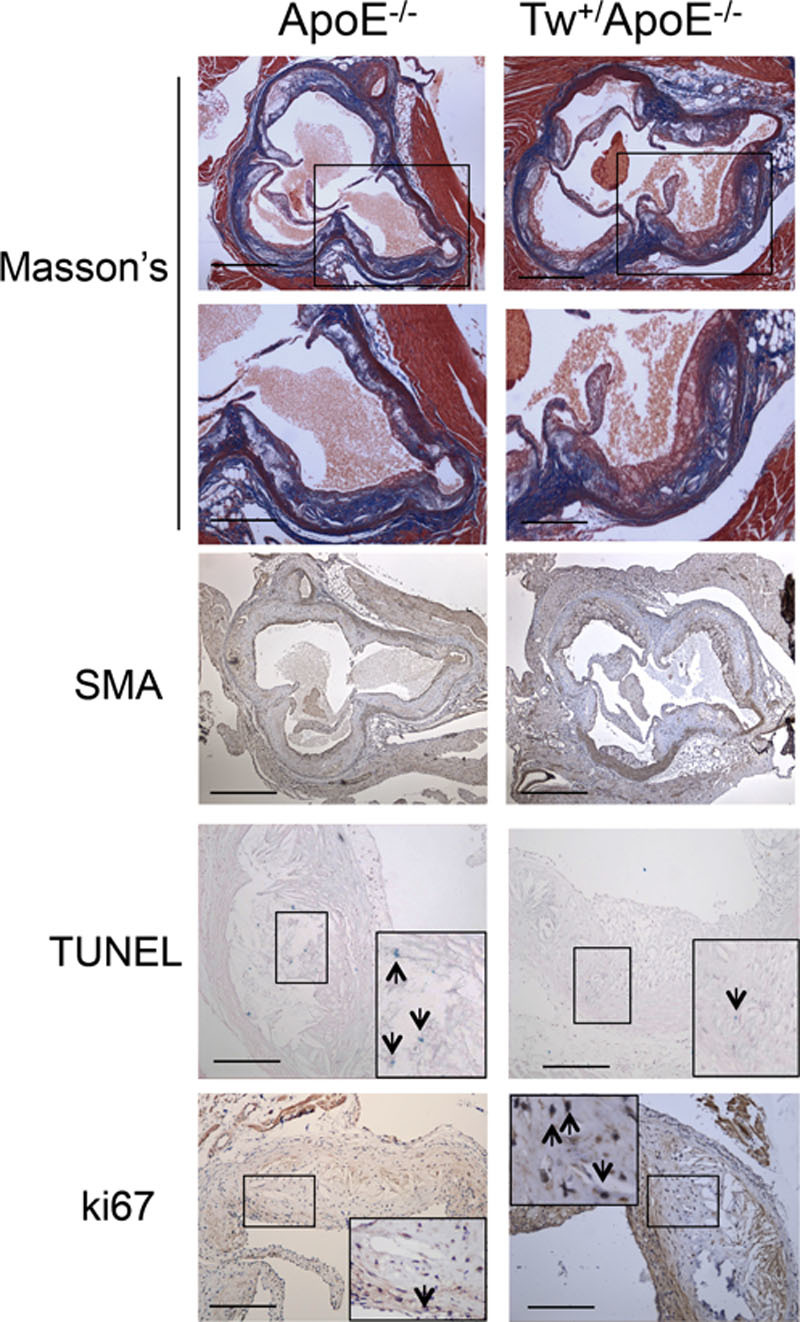
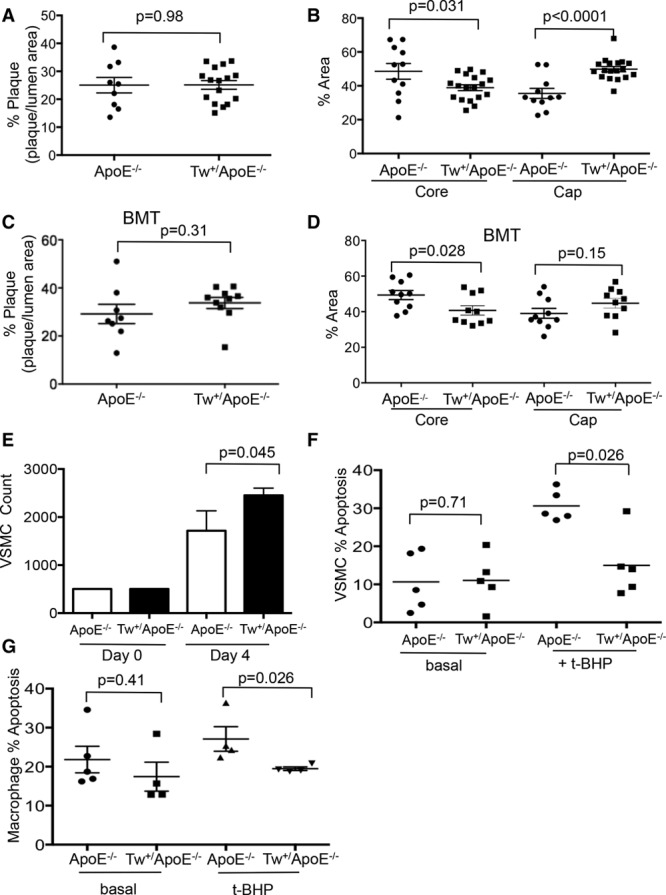
Approach and results: Human atherosclerotic plaques showed marked mitochondrial dysfunction, manifested as reduced mtDNA copy number and oxygen consumption rate in fibrous cap and core regions. Vascular smooth muscle cells derived from plaques showed impaired mitochondrial respiration, reduced complex I expression, and increased mitophagy, which was induced by oxidized low-density lipoprotein. Apolipoprotein E-deficient (ApoE-/-) mice showed decreased mtDNA integrity and mitochondrial respiration, associated with increased mitochondrial reactive oxygen species. To determine whether alleviating mtDNA damage and increasing mitochondrial respiration affects atherogenesis, we studied ApoE-/- mice overexpressing the mitochondrial helicase Twinkle (Tw+/ApoE-/-). Tw+/ApoE-/- mice showed increased mtDNA integrity, copy number, respiratory complex abundance, and respiration. Tw+/ApoE-/- mice had decreased necrotic core and increased fibrous cap areas, and Tw+/ApoE-/- bone marrow transplantation also reduced core areas. Twinkle increased vascular smooth muscle cell mtDNA integrity and respiration. Twinkle also promoted vascular smooth muscle cell proliferation and protected both vascular smooth muscle cells and macrophages from oxidative stress-induced apoptosis.
Conclusions: Endogenous mtDNA damage in mouse and human atherosclerosis is associated with significantly reduced mitochondrial respiration. Reducing mtDNA damage and increasing mitochondrial respiration decrease necrotic core and increase fibrous cap areas independently of changes in reactive oxygen species and may be a promising therapeutic strategy in atherosclerosis.
Keywords: atherosclerosis; mitochondria; reactive oxygen species; respiration; vascular smooth muscle.
© 2017 The Authors.
Figures
Comment in
-
Mitochondrial Respiration and Atherosclerosis: R-E-S-P-I-R-E. Find Out What it Means to Mϕ (and VSMC).Arterioscler Thromb Vasc Biol. 2017 Dec;37(12):2229-2230. doi: 10.1161/ATVBAHA.117.310298. Arterioscler Thromb Vasc Biol. 2017. PMID: 29162598 Free PMC article. No abstract available.
Similar articles
-
Mitochondrial DNA damage can promote atherosclerosis independently of reactive oxygen species through effects on smooth muscle cells and monocytes and correlates with higher-risk plaques in humans.Circulation. 2013 Aug 13;128(7):702-12. doi: 10.1161/CIRCULATIONAHA.113.002271. Epub 2013 Jul 10. Circulation. 2013. PMID: 23841983
-
Attenuated Superoxide Dismutase 2 Activity Induces Atherosclerotic Plaque Instability During Aging in Hyperlipidemic Mice.J Am Heart Assoc. 2017 Oct 27;6(11):e006775. doi: 10.1161/JAHA.117.006775. J Am Heart Assoc. 2017. PMID: 29079564 Free PMC article.
-
METTL4-Mediated Mitochondrial DNA N6-Methyldeoxyadenosine Promoting Macrophage Inflammation and Atherosclerosis.Circulation. 2025 Apr;151(13):946-965. doi: 10.1161/CIRCULATIONAHA.124.069574. Epub 2024 Dec 17. Circulation. 2025. PMID: 39687989 Free PMC article.
-
Mitochondrion as a Selective Target for the Treatment of Atherosclerosis: Role of Mitochondrial DNA Mutations and Defective Mitophagy in the Pathogenesis of Atherosclerosis and Chronic Inflammation.Curr Neuropharmacol. 2020;18(11):1064-1075. doi: 10.2174/1570159X17666191118125018. Curr Neuropharmacol. 2020. PMID: 31744449 Free PMC article. Review.
-
Vascular smooth muscle cell death, autophagy and senescence in atherosclerosis.Cardiovasc Res. 2018 Mar 15;114(4):622-634. doi: 10.1093/cvr/cvy007. Cardiovasc Res. 2018. PMID: 29360955 Review.
Cited by
-
Altered mitochondrial quality control in Atg7-deficient VSMCs promotes enhanced apoptosis and is linked to unstable atherosclerotic plaque phenotype.Cell Death Dis. 2019 Feb 11;10(2):119. doi: 10.1038/s41419-019-1400-0. Cell Death Dis. 2019. PMID: 30741928 Free PMC article.
-
Age-associated adipose tissue inflammation promotes monocyte chemotaxis and enhances atherosclerosis.Aging Cell. 2023 Feb;22(2):e13783. doi: 10.1111/acel.13783. Epub 2023 Jan 23. Aging Cell. 2023. PMID: 36683460 Free PMC article.
-
Response to: Comment on "Role of Mitochondrial Genome Mutations in Pathogenesis of Carotid Atherosclerosis".Oxid Med Cell Longev. 2018 Aug 9;2018:7620234. doi: 10.1155/2018/7620234. eCollection 2018. Oxid Med Cell Longev. 2018. PMID: 30159117 Free PMC article. No abstract available.
-
Uric Acid in Inflammation and the Pathogenesis of Atherosclerosis.Int J Mol Sci. 2021 Nov 17;22(22):12394. doi: 10.3390/ijms222212394. Int J Mol Sci. 2021. PMID: 34830282 Free PMC article. Review.
-
Macrophage immunometabolism in diabetes-associated atherosclerosis.Immunometabolism (Cobham). 2023 Oct 16;5(4):e00032. doi: 10.1097/IN9.0000000000000032. eCollection 2023 Oct. Immunometabolism (Cobham). 2023. PMID: 37849988 Free PMC article. Review.
References
-
- Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, Schreier PH, Smith AJ, Staden R, Young IG. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–465. - PubMed
-
- Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly-Y M, Gidlöf S, Oldfors A, Wibom R, Törnell J, Jacobs HT, Larsson NG. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–423. doi: 10.1038/nature02517. - PubMed
-
- Tyynismaa H, Sembongi H, Bokori-Brown M, Granycome C, Ashley N, Poulton J, Jalanko A, Spelbrink JN, Holt IJ, Suomalainen A. Twinkle helicase is essential for mtDNA maintenance and regulates mtDNA copy number. Hum Mol Genet. 2004;13:3219–3227. doi: 10.1093/hmg/ddh342. - PubMed
MeSH terms
Substances
Grants and funding
- FS/10/70/28507/BHF_/British Heart Foundation/United Kingdom
- CH/2000003/12800/BHF_/British Heart Foundation/United Kingdom
- PG/13/25/30014/BHF_/British Heart Foundation/United Kingdom
- PG/14/69/31032/BHF_/British Heart Foundation/United Kingdom
- PG/16/63/32307/BHF_/British Heart Foundation/United Kingdom
- MC_U105663142/MRC_/Medical Research Council/United Kingdom
- PG/16/11/32021/BHF_/British Heart Foundation/United Kingdom
- 110158/Z/15/Z/WT_/Wellcome Trust/United Kingdom
- PG/11/57/29003/BHF_/British Heart Foundation/United Kingdom
- 110159/Z/15/Z/WT_/Wellcome Trust/United Kingdom
- RG/13/14/30314/BHF_/British Heart Foundation/United Kingdom
- MC_UU_00015/3/MRC_/Medical Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
