

Heat Shock Protein 70 (Hsp70) Suppresses RIP1-Dependent Apoptotic and Necroptotic Cascades
- PMID: 28970360
- PMCID: PMC6101036
- DOI: 10.1158/1541-7786.MCR-17-0408
Heat Shock Protein 70 (Hsp70) Suppresses RIP1-Dependent Apoptotic and Necroptotic Cascades
Abstract
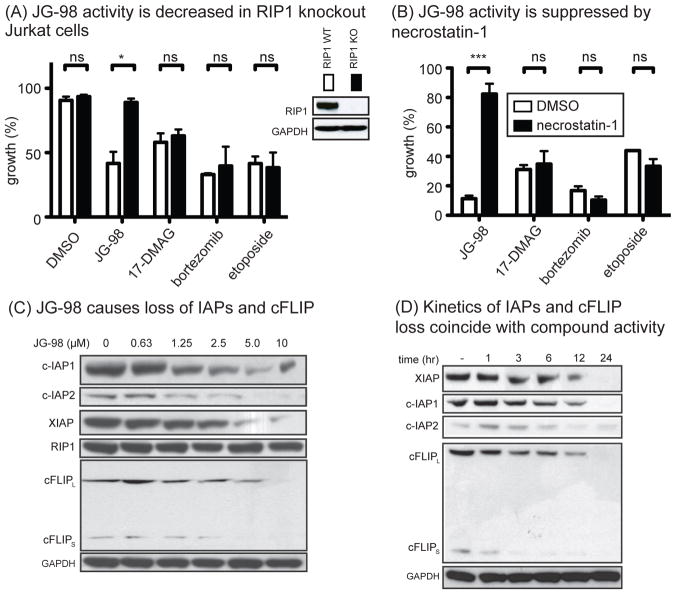
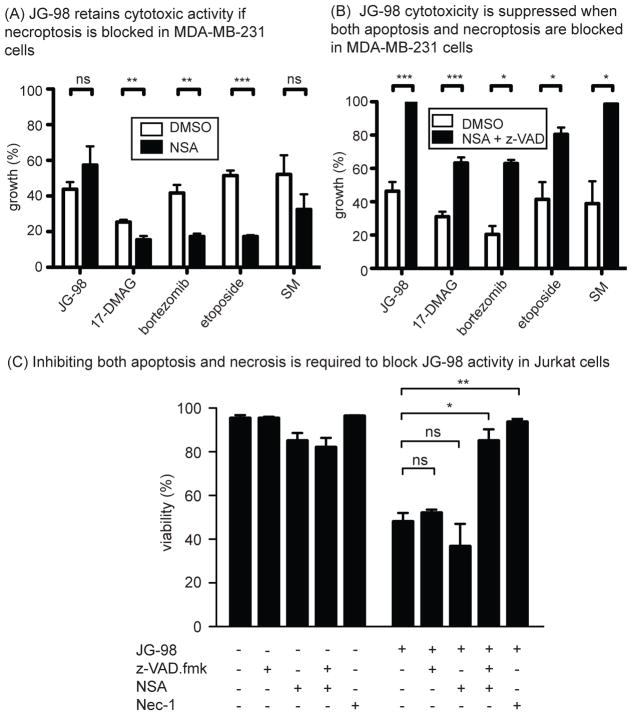
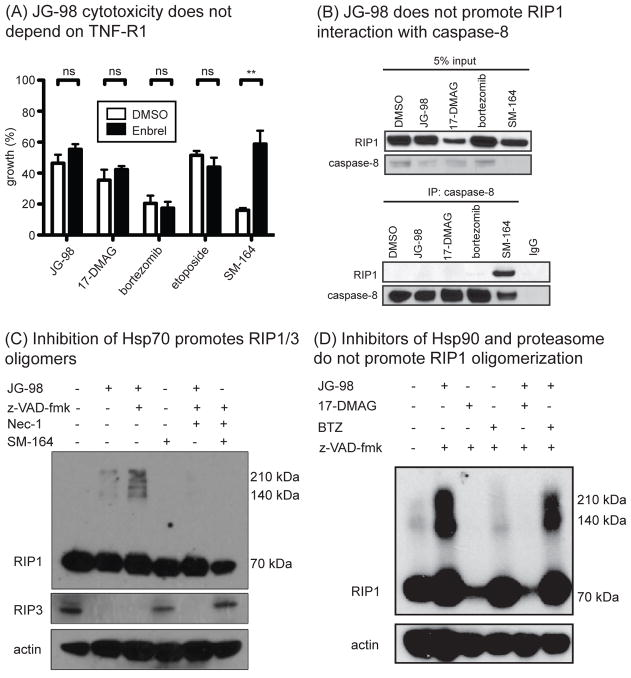
Hsp70 is a molecular chaperone that binds to "client" proteins and protects them from protein degradation. Hsp70 is essential for the survival of many cancer cells, but it is not yet clear which of its clients are involved. Using structurally distinct chemical inhibitors, we found that many of the well-known clients of the related chaperone, Hsp90, are not strikingly responsive to Hsp70 inhibition. Rather, Hsp70 appeared to be important for the stability of the RIP1 (RIPK1) regulators: cIAP1/2 (BIRC1 and BIRC3), XIAP, and cFLIPS/L (CFLAR). These results suggest that Hsp70 limits apoptosis and necroptosis pathways downstream of RIP1. Consistent with this model, MDA-MB-231 breast cancer cells treated with Hsp70 inhibitors underwent apoptosis, while cotreatment with z-VAD.fmk switched the cell death pathway to necroptosis. In addition, cell death in response to Hsp70 inhibitors was strongly suppressed by RIP1 knockdown or inhibitors. Thus, these data indicate that Hsp70 plays a previously unrecognized and important role in suppressing RIP1 activity.Implications: These findings clarify the role of Hsp70 in prosurvival signaling and suggest IAPs as potential new biomarkers for Hsp70 inhibition. Mol Cancer Res; 16(1); 58-68. ©2017 AACR.
©2017 American Association for Cancer Research.
Conflict of interest statement
The authors claim no competing financial interests.
Figures
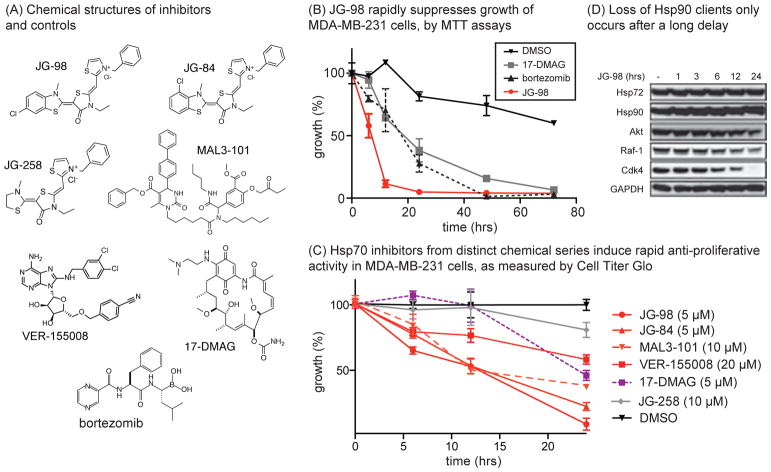
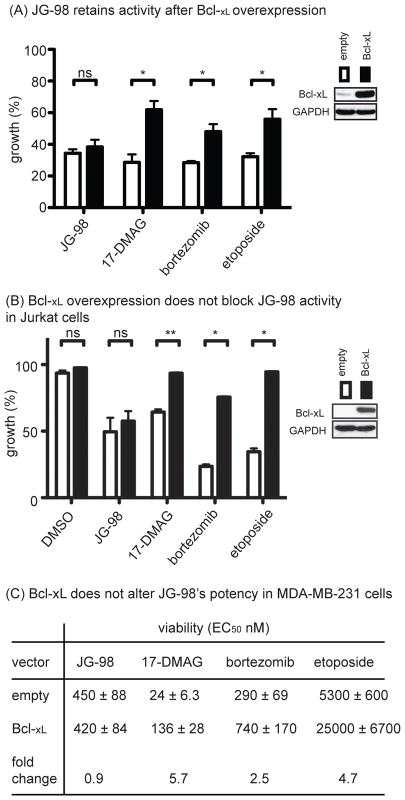
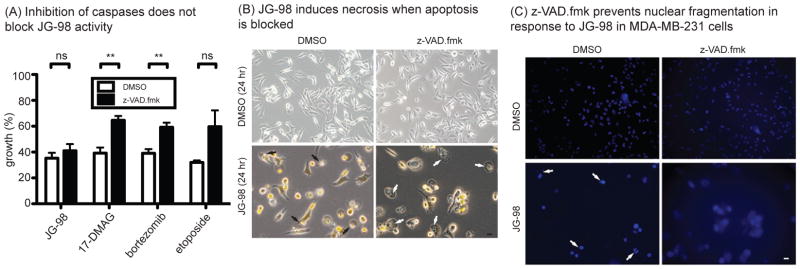
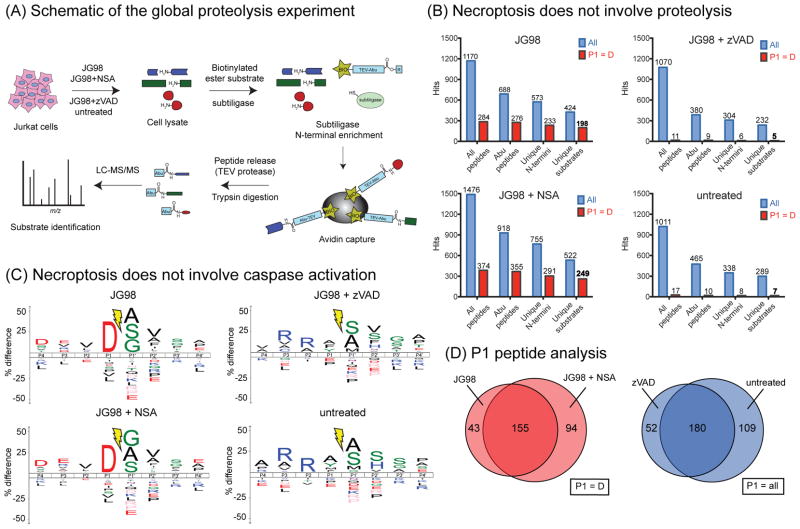
Similar articles
-
Non-benzoquinone geldanamycin analogs trigger various forms of death in human breast cancer cells.J Exp Clin Cancer Res. 2016 Sep 22;35(1):149. doi: 10.1186/s13046-016-0428-6. J Exp Clin Cancer Res. 2016. PMID: 27658586 Free PMC article.
-
X-linked inhibitor of apoptosis protein (XIAP) is a client of heat shock protein 70 (Hsp70) and a biomarker of its inhibition.J Biol Chem. 2018 Feb 16;293(7):2370-2380. doi: 10.1074/jbc.RA117.000634. Epub 2017 Dec 18. J Biol Chem. 2018. PMID: 29255093 Free PMC article.
-
Cellular IAP proteins and LUBAC differentially regulate necrosome-associated RIP1 ubiquitination.Cell Death Dis. 2015 Jun 25;6(6):e1800. doi: 10.1038/cddis.2015.158. Cell Death Dis. 2015. PMID: 26111062 Free PMC article.
-
RIP1 post-translational modifications.Biochem J. 2022 May 13;479(9):929-951. doi: 10.1042/BCJ20210725. Biochem J. 2022. PMID: 35522161 Review.
-
Control of life-or-death decisions by RIP1 kinase.Annu Rev Physiol. 2014;76:129-50. doi: 10.1146/annurev-physiol-021113-170259. Epub 2013 Sep 20. Annu Rev Physiol. 2014. PMID: 24079414 Review.
Cited by
-
Hydrogen Sulfide Gas Exposure Induces Necroptosis and Promotes Inflammation through the MAPK/NF-κB Pathway in Broiler Spleen.Oxid Med Cell Longev. 2019 Jul 31;2019:8061823. doi: 10.1155/2019/8061823. eCollection 2019. Oxid Med Cell Longev. 2019. PMID: 31467636 Free PMC article.
-
Cellular protection from H2O2 toxicity by Fv-Hsp70: protection via catalase and gamma-glutamyl-cysteine synthase.Cell Stress Chaperones. 2023 Jul;28(4):429-439. doi: 10.1007/s12192-023-01349-6. Epub 2023 May 12. Cell Stress Chaperones. 2023. PMID: 37171750 Free PMC article.
-
Heat Shock Protein Family A Member 1A Attenuates Apoptosis and Oxidative Stress via ERK/JNK Pathway in Hyperplastic Prostate.MedComm (2020). 2025 Mar 10;6(3):e70129. doi: 10.1002/mco2.70129. eCollection 2025 Mar. MedComm (2020). 2025. PMID: 40066224 Free PMC article.
-
The protein phosphatase-2A subunit PR130 is involved in the formation of cytotoxic protein aggregates in pancreatic ductal adenocarcinoma cells.Cell Commun Signal. 2024 Apr 3;22(1):217. doi: 10.1186/s12964-024-01597-8. Cell Commun Signal. 2024. PMID: 38570831 Free PMC article.
-
Adhesion-mediated mechanosignaling forces mitohormesis.Cell Metab. 2021 Jul 6;33(7):1322-1341.e13. doi: 10.1016/j.cmet.2021.04.017. Epub 2021 May 20. Cell Metab. 2021. PMID: 34019840 Free PMC article.
References
-
- Nylandsted J, Brand K, Jaattela M. Heat shock protein 70 is required for the survival of cancer cells. Ann N Y Acad Sci. 2000;926:122–125. - PubMed
-
- Nanbu K, et al. Prognostic significance of heat shock proteins HSP70 and HSP90 in endometrial carcinomas. Cancer detection and prevention. 1998;22:549–555. - PubMed
-
- Brodsky JL, Chiosis G. Hsp70 molecular chaperones: emerging roles in human disease and identification of small molecule modulators. Curr Top Med Chem. 2006;6:1215–1225. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
