

Multiple regulatory mechanisms of the biological function of NRF3 (NFE2L3) control cancer cell proliferation
- PMID: 28970512
- PMCID: PMC5624902
- DOI: 10.1038/s41598-017-12675-y
Multiple regulatory mechanisms of the biological function of NRF3 (NFE2L3) control cancer cell proliferation
Abstract
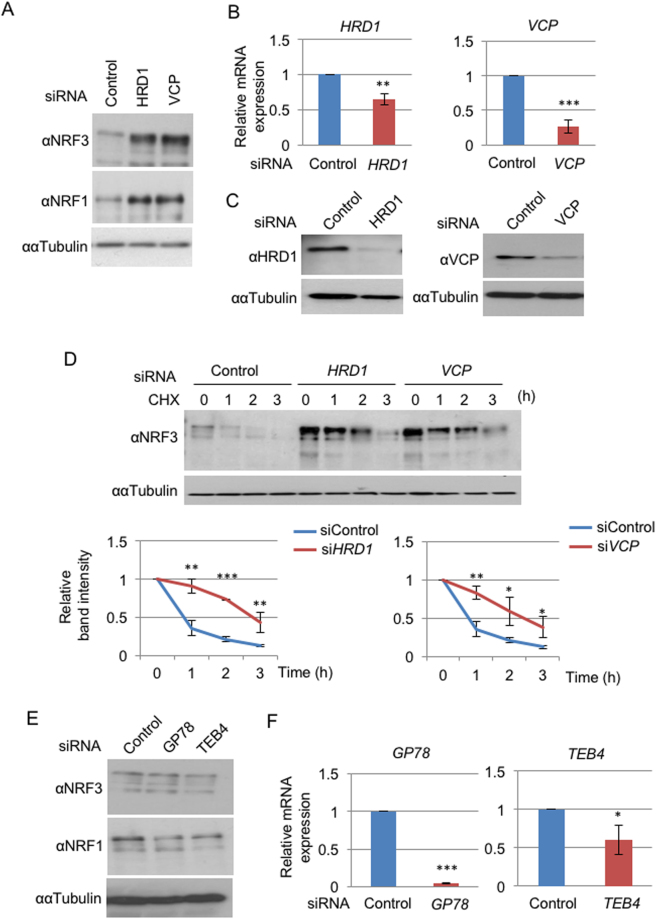
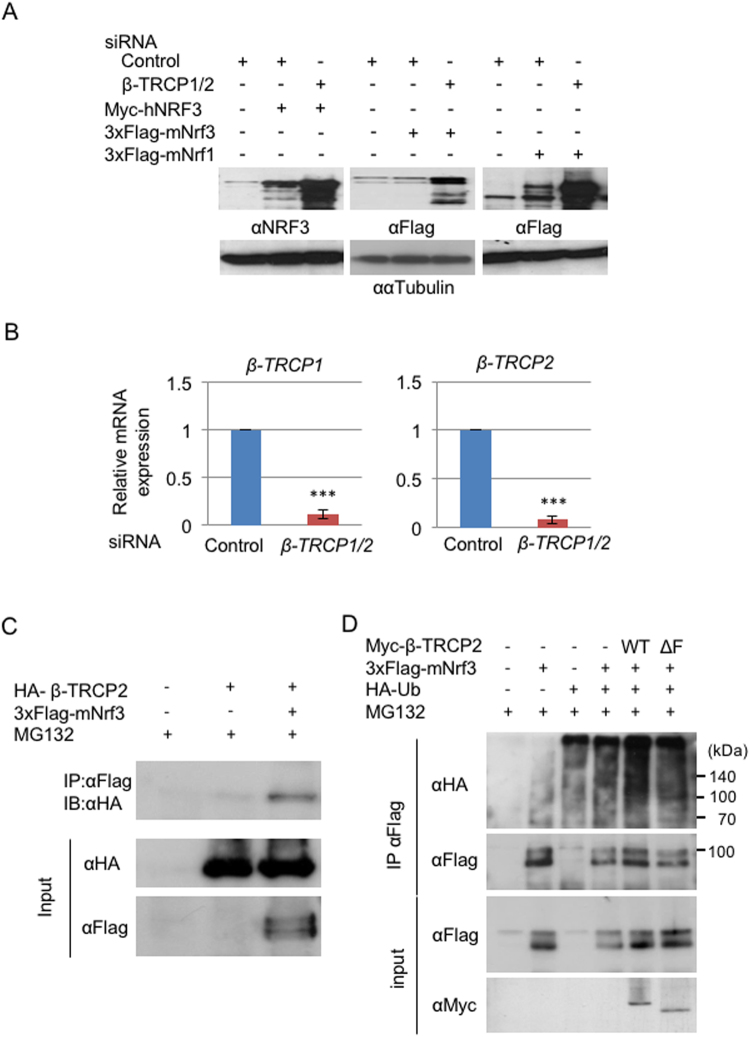
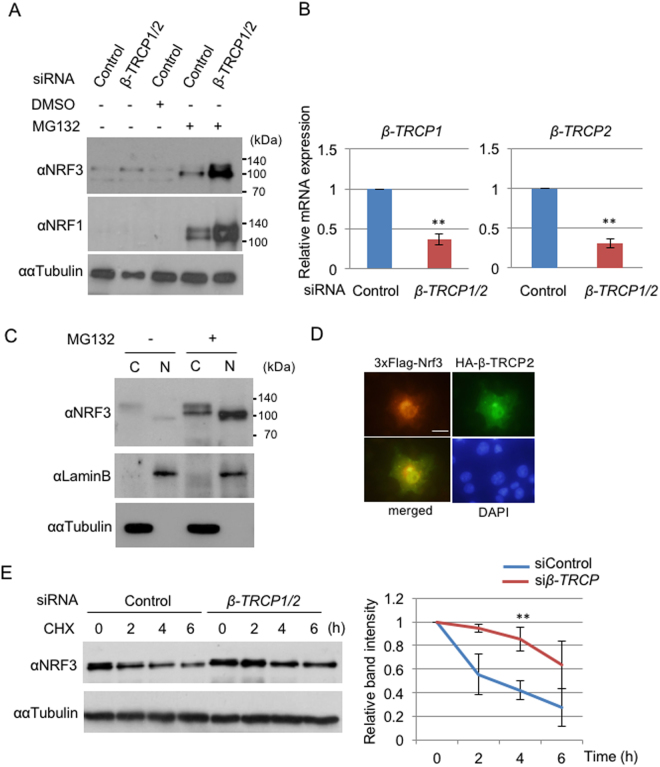
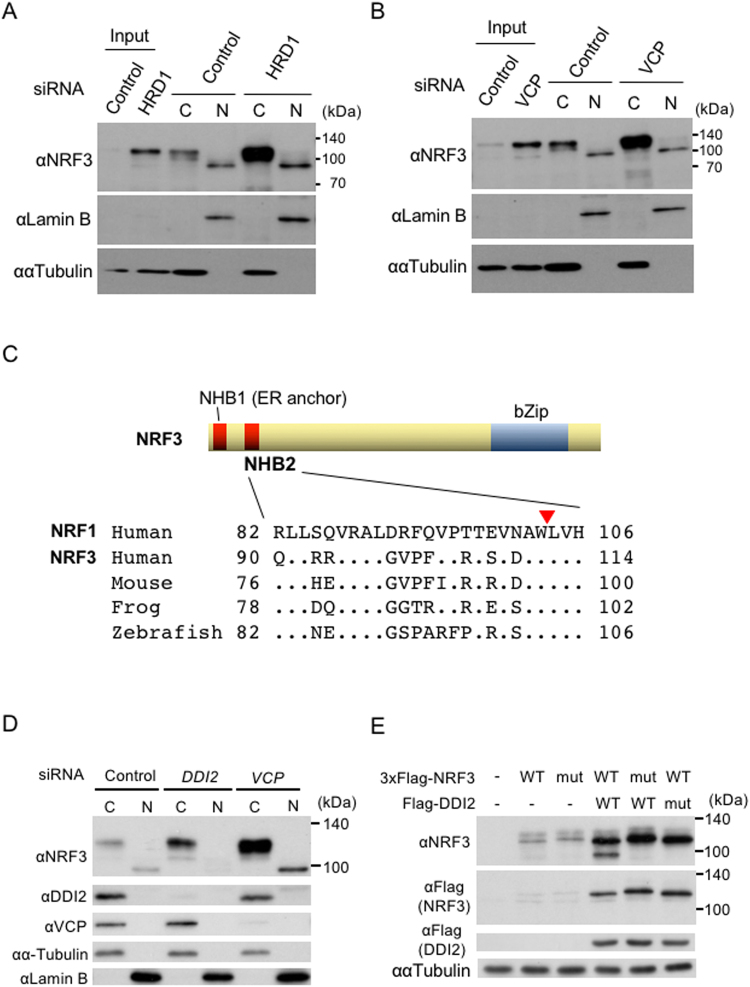
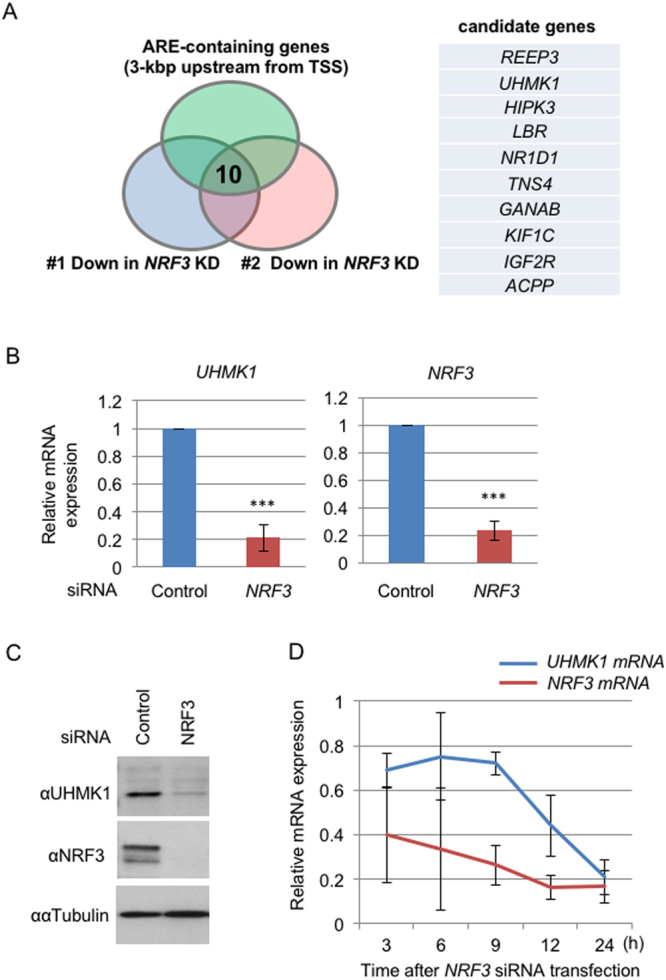
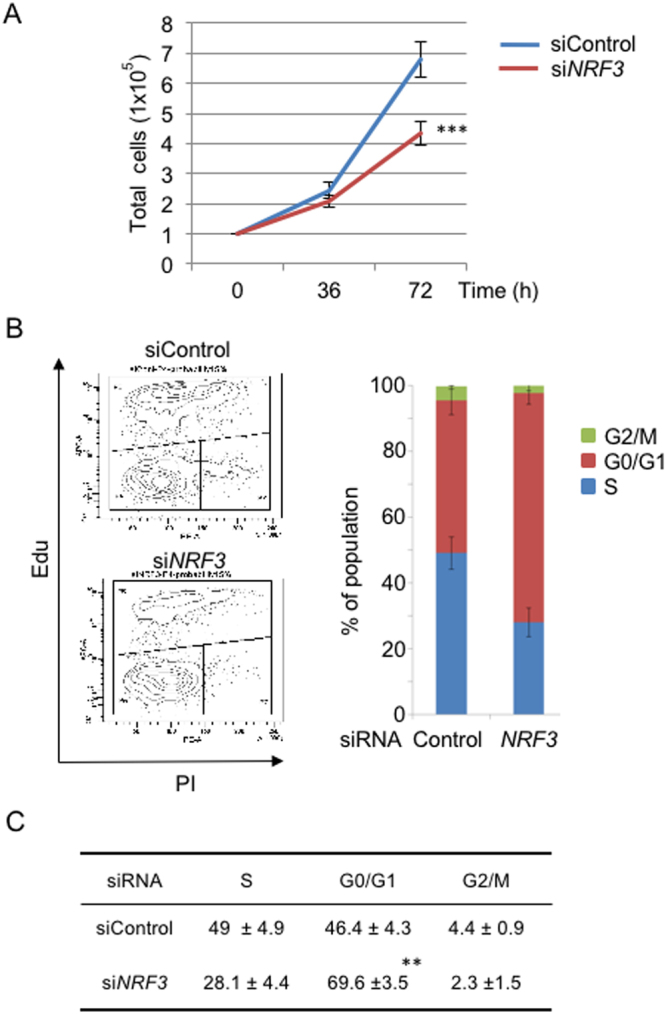
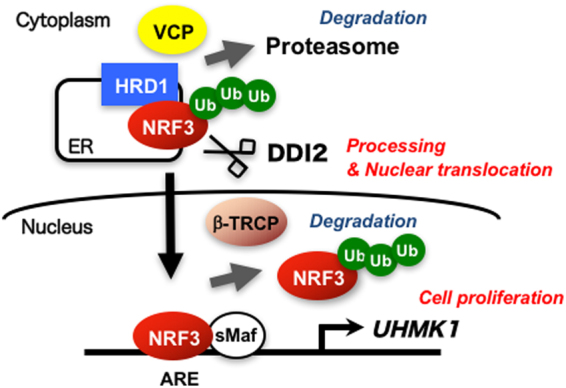
Accumulated evidence suggests a physiological relationship between the transcription factor NRF3 (NFE2L3) and cancers. Under physiological conditions, NRF3 is repressed by its endoplasmic reticulum (ER) sequestration. In response to unidentified signals, NRF3 enters the nucleus and modulates gene expression. However, molecular mechanisms underlying the nuclear translocation of NRF3 and its target gene in cancer cells remain poorly understood. We herein report that multiple regulation of NRF3 activities controls cell proliferation. Our analyses reveal that under physiological conditions, NRF3 is rapidly degraded by the ER-associated degradation (ERAD) ubiquitin ligase HRD1 and valosin-containing protein (VCP) in the cytoplasm. Furthermore, NRF3 is also degraded by β-TRCP, an adaptor for the Skp1-Cul1-F-box protein (SCF) ubiquitin ligase in the nucleus. The nuclear translocation of NRF3 from the ER requires the aspartic protease DNA-damage inducible 1 homolog 2 (DDI2) but does not require inhibition of its HRD1-VCP-mediated degradation. Finally, NRF3 mediates gene expression of the cell cycle regulator U2AF homology motif kinase 1 (UHMK1) for cell proliferation. Collectively, our study provides us many insights into the molecular regulation and biological function of NRF3 in cancer cells.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
