

TRAF3IP2 mediates high glucose-induced endothelin-1 production as well as endothelin-1-induced inflammation in endothelial cells
- PMID: 28971844
- PMCID: PMC5866390
- DOI: 10.1152/ajpheart.00478.2017
TRAF3IP2 mediates high glucose-induced endothelin-1 production as well as endothelin-1-induced inflammation in endothelial cells
Abstract
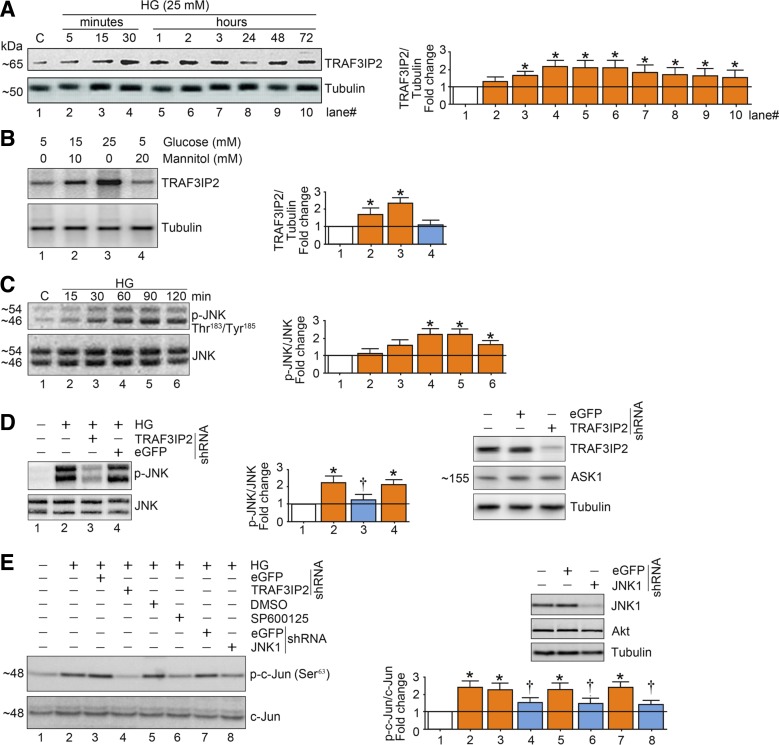
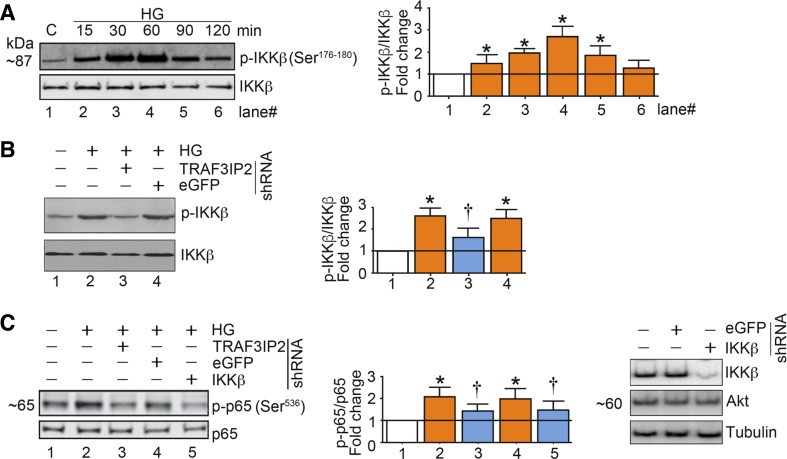
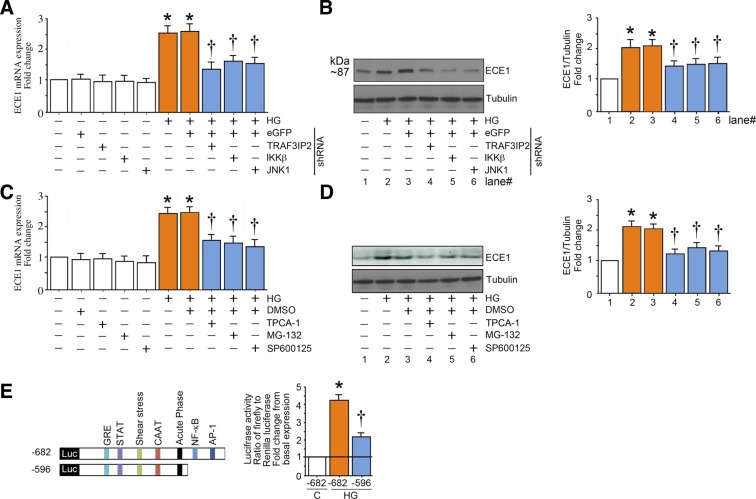
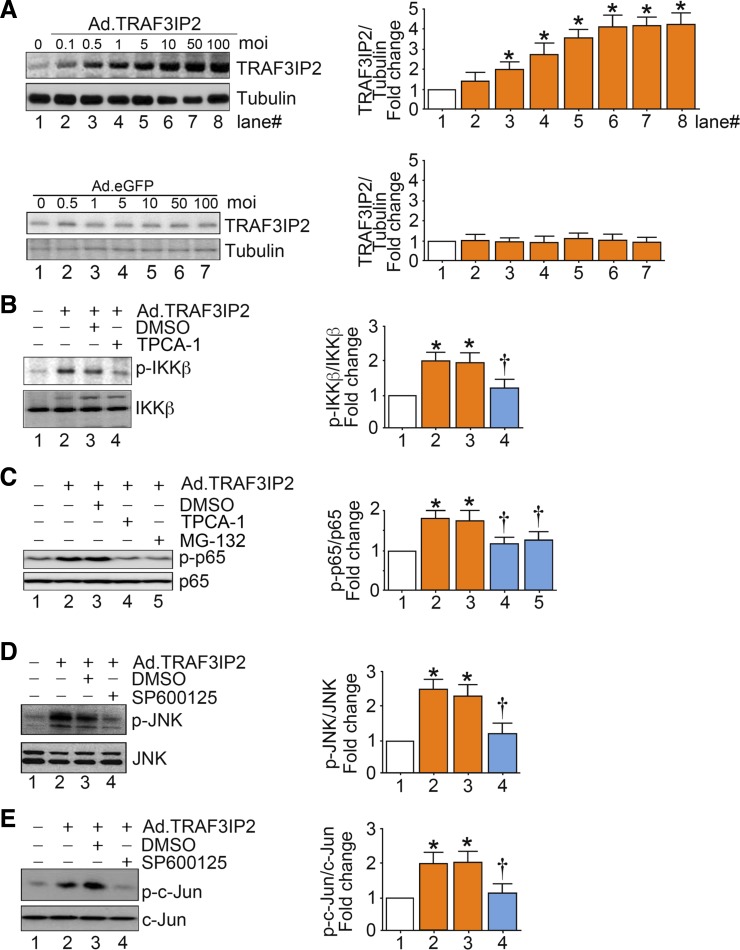
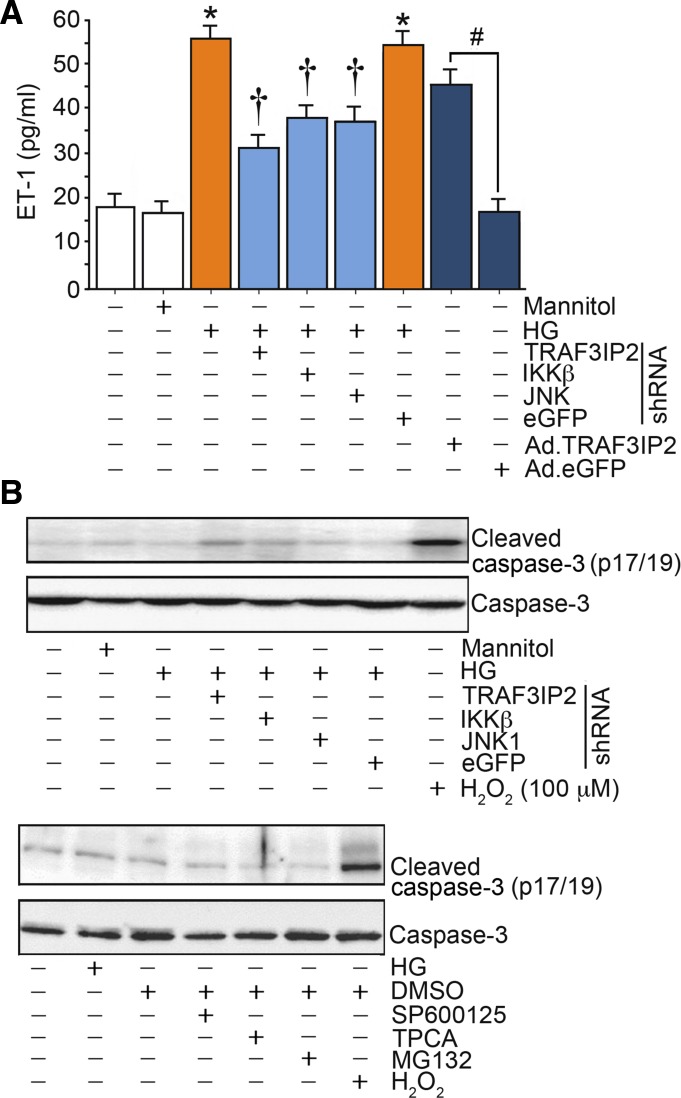
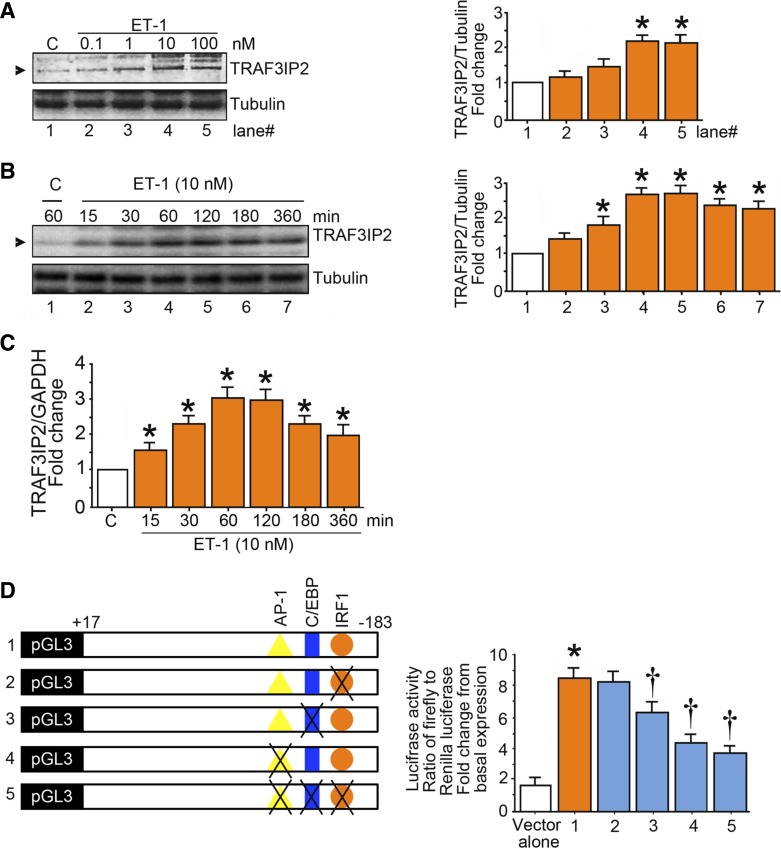
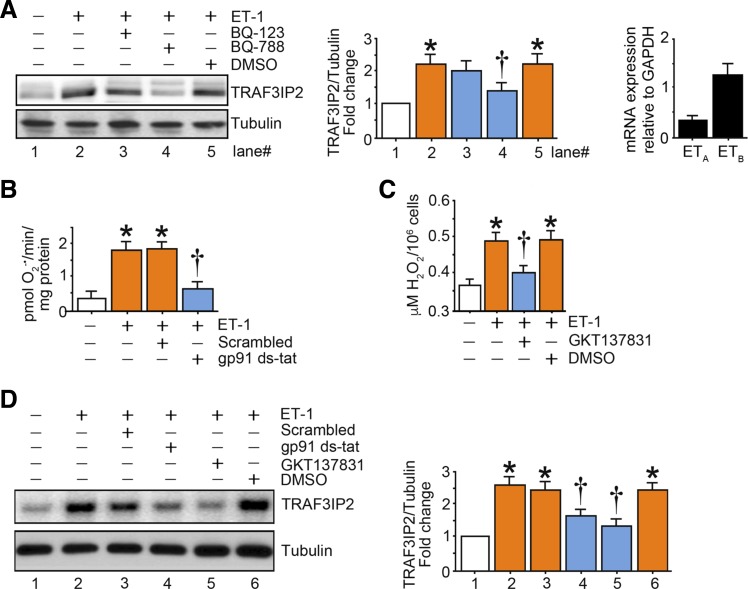
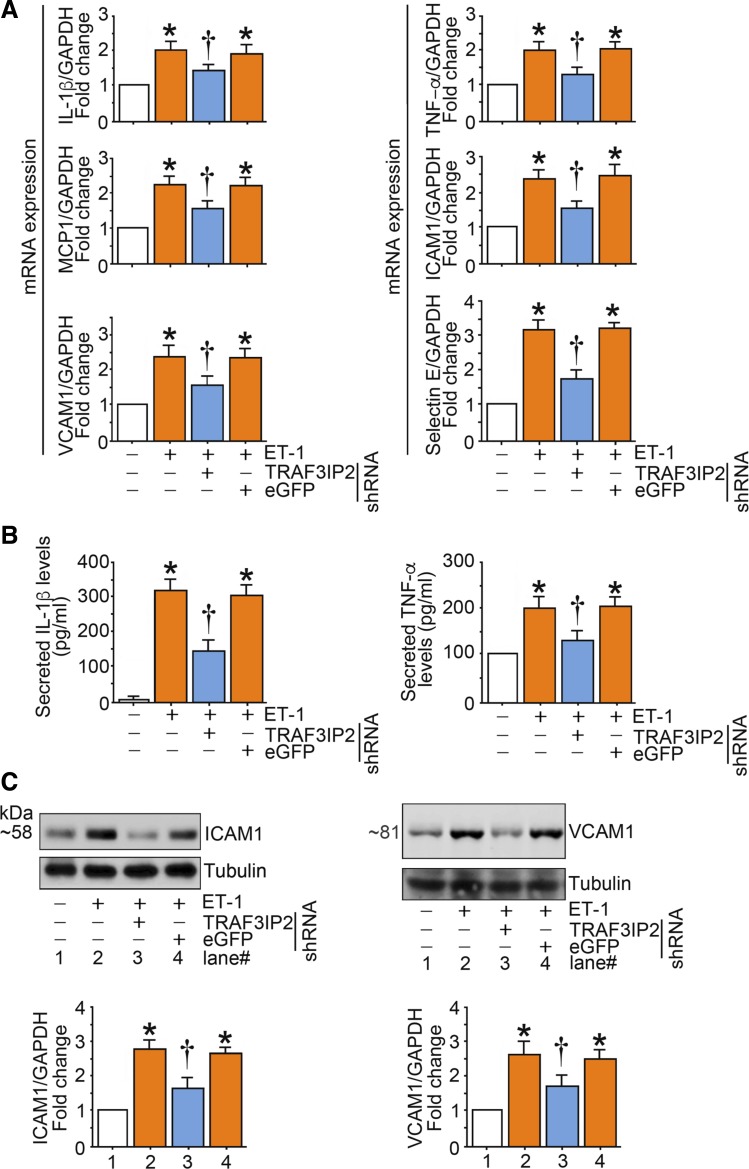
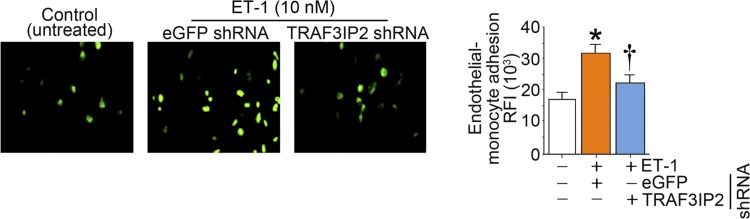
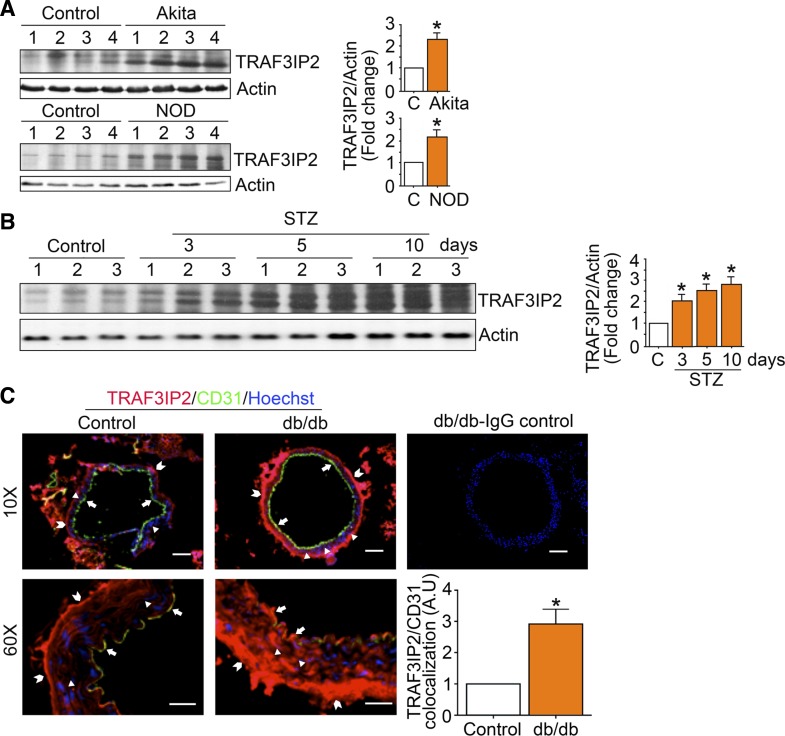
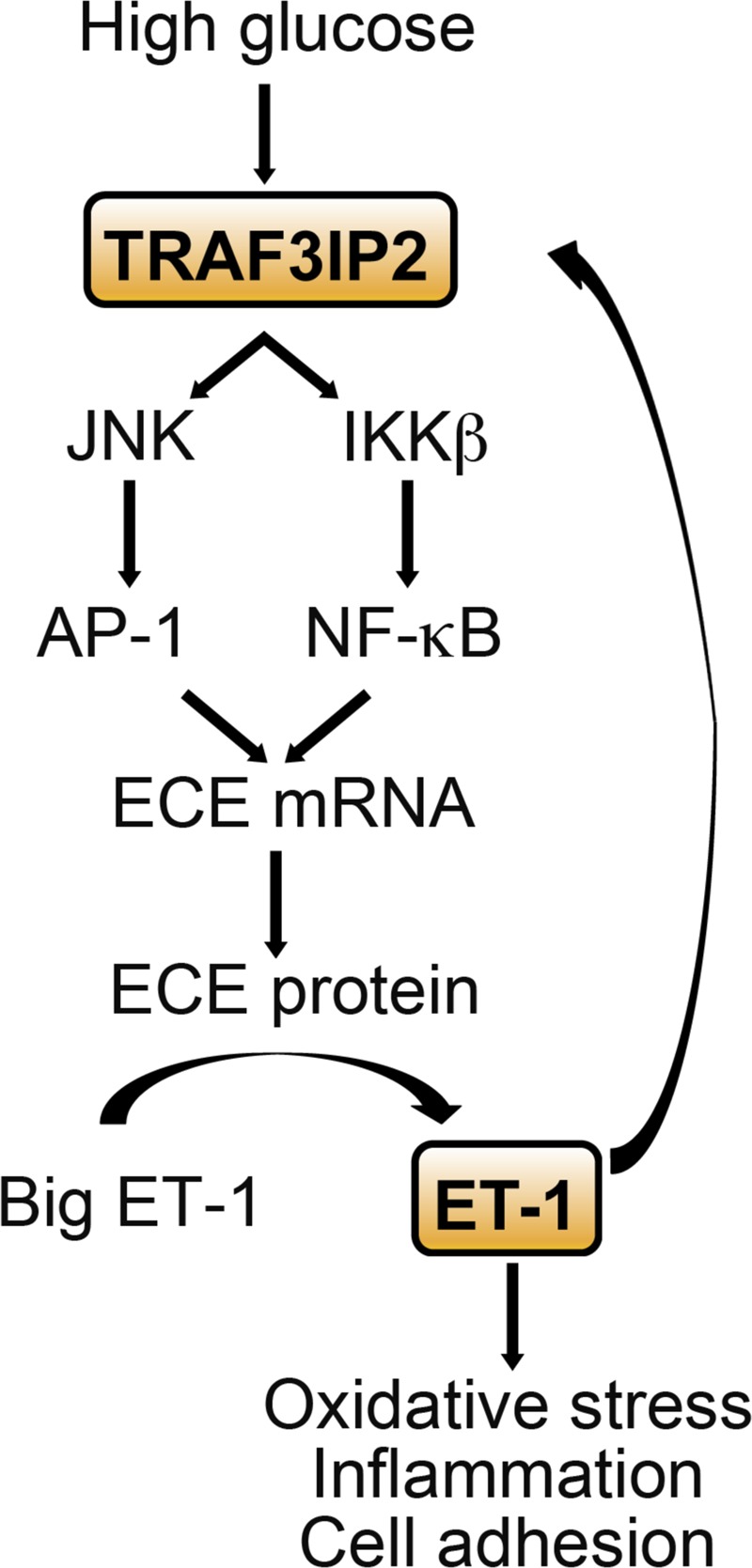
Hyperglycemia-induced production of endothelin (ET)-1 is a hallmark of endothelial dysfunction in diabetes. Although the detrimental vascular effects of increased ET-1 are well known, the molecular mechanisms regulating endothelial synthesis of ET-1 in the setting of diabetes remain largely unidentified. Here, we show that adapter molecule TRAF3 interacting protein 2 (TRAF3IP2) mediates high glucose-induced ET-1 production in endothelial cells and ET-1-mediated endothelial cell inflammation. Specifically, we found that high glucose upregulated TRAF3IP2 in human aortic endothelial cells, which subsequently led to activation of JNK and IKKβ. shRNA-mediated silencing of TRAF3IP2, JNK1, or IKKβ abrogated high-glucose-induced ET-converting enzyme 1 expression and ET-1 production. Likewise, overexpression of TRAF3IP2, in the absence of high glucose, led to activation of JNK and IKKβ as well as increased ET-1 production. Furthermore, ET-1 transcriptionally upregulated TRAF3IP2, and this upregulation was prevented by pharmacological inhibition of ET-1 receptor B using BQ-788, or inhibition of NADPH oxidase-derived reactive oxygen species using gp91ds-tat and GKT137831. Notably, we found that knockdown of TRAF3IP2 abolished ET-1-induced proinflammatory and adhesion molecule (IL-1β, TNF-α, monocyte chemoattractant protein 1, ICAM-1, VCAM-1, and E-selectin) expression and monocyte adhesion to endothelial cells. Finally, we report that TRAF3IP2 is upregulated and colocalized with CD31, an endothelial marker, in the aorta of diabetic mice. Collectively, findings from the present study identify endothelial TRAF3IP2 as a potential new therapeutic target to suppress ET-1 production and associated vascular complications in diabetes. NEW & NOTEWORTHY This study provides the first evidence that the adapter molecule TRAF3 interacting protein 2 mediates high glucose-induced production of endothelin-1 by endothelial cells as well as endothelin-1-mediated endothelial cell inflammation. The findings presented herein suggest that TRAF3 interacting protein 2 may be an important therapeutic target in diabetic vasculopathy characterized by excess endothelin-1 production.
Keywords: TRAF3 interacting protein 2; endothelial dysfunction; endothelin-1; hyperglycemia.
Figures
Comment in
-
Too much TRAFfic at the crossroads of diabetes and endothelial dysfunction.Am J Physiol Heart Circ Physiol. 2018 Jan 1;314(1):H65-H67. doi: 10.1152/ajpheart.00614.2017. Epub 2017 Nov 3. Am J Physiol Heart Circ Physiol. 2018. PMID: 29101184 Free PMC article. No abstract available.
Similar articles
-
CIKS (Act1 or TRAF3IP2) mediates high glucose-induced endothelial dysfunction.Cell Signal. 2013 Jan;25(1):359-71. doi: 10.1016/j.cellsig.2012.10.009. Epub 2012 Oct 17. Cell Signal. 2013. PMID: 23085260 Free PMC article.
-
OxLDL induces endothelial dysfunction and death via TRAF3IP2: inhibition by HDL3 and AMPK activators.Free Radic Biol Med. 2014 May;70:117-28. doi: 10.1016/j.freeradbiomed.2014.02.014. Epub 2014 Feb 20. Free Radic Biol Med. 2014. PMID: 24561578 Free PMC article.
-
TRAF3IP2 (TRAF3 Interacting Protein 2) Mediates Obesity-Associated Vascular Insulin Resistance and Dysfunction in Male Mice.Hypertension. 2020 Oct;76(4):1319-1329. doi: 10.1161/HYPERTENSIONAHA.120.15262. Epub 2020 Aug 24. Hypertension. 2020. PMID: 32829657 Free PMC article.
-
c-Src tyrosine kinase mediates high glucose-induced endothelin-1 expression.Int J Biochem Cell Biol. 2016 Jun;75:123-30. doi: 10.1016/j.biocel.2016.04.008. Epub 2016 Apr 19. Int J Biochem Cell Biol. 2016. PMID: 27102411
-
Clinical efficacies, underlying mechanisms and molecular targets of Chinese medicines for diabetic nephropathy treatment and management.Acta Pharm Sin B. 2021 Sep;11(9):2749-2767. doi: 10.1016/j.apsb.2020.12.020. Epub 2021 Feb 2. Acta Pharm Sin B. 2021. PMID: 34589395 Free PMC article. Review.
Cited by
-
New insights into mechanisms of endothelial insulin resistance in type 2 diabetes.Am J Physiol Heart Circ Physiol. 2022 Dec 1;323(6):H1231-H1238. doi: 10.1152/ajpheart.00537.2022. Epub 2022 Nov 4. Am J Physiol Heart Circ Physiol. 2022. PMID: 36331555 Free PMC article. Review.
-
Glucose-induced oxidative stress and accelerated aging in endothelial cells are mediated by the depletion of mitochondrial SIRTs.Physiol Rep. 2020 Feb;8(3):e14331. doi: 10.14814/phy2.14331. Physiol Rep. 2020. PMID: 32026628 Free PMC article.
-
Molecular mechanisms and genetic regulation in atherosclerosis.Int J Cardiol Heart Vasc. 2018 Sep 25;21:36-44. doi: 10.1016/j.ijcha.2018.09.006. eCollection 2018 Dec. Int J Cardiol Heart Vasc. 2018. PMID: 30276232 Free PMC article. Review.
-
Uncoupled nitric oxide synthase activity promotes colorectal cancer progression.Front Oncol. 2023 Mar 14;13:1165326. doi: 10.3389/fonc.2023.1165326. eCollection 2023. Front Oncol. 2023. PMID: 36998441 Free PMC article.
-
Empagliflozin Reverses Oxidized LDL-Induced RECK Suppression, Cardiotrophin-1 Expression, MMP Activation, and Human Aortic Smooth Muscle Cell Proliferation and Migration.Mediators Inflamm. 2023 Oct 4;2023:6112301. doi: 10.1155/2023/6112301. eCollection 2023. Mediators Inflamm. 2023. PMID: 37830075 Free PMC article.
References
-
- Adamopoulos C, Piperi C, Gargalionis AN, Dalagiorgou G, Spilioti E, Korkolopoulou P, Diamanti-Kandarakis E, Papavassiliou AG. Advanced glycation end products upregulate lysyl oxidase and endothelin-1 in human aortic endothelial cells via parallel activation of ERK1/2-NF-kappaB and JNK-AP-1 signaling pathways. Cell Mol Life Sci 73: 1685–1698, 2016. doi:10.1007/s00018-015-2091-z. - DOI - PMC - PubMed
-
- Aroor AR, Habibi J, Kandikattu HK, Garro-Kacher M, Barron B, Chen D, Hayden MR, Whaley-Connell A, Bender SB, Klein T, Padilla J, Sowers JR, Chandrasekar B, DeMarco VG. Dipeptidyl peptidase-4 (DPP-4) inhibition with linagliptin reduces western diet-induced myocardial TRAF3IP2 expression, inflammation and fibrosis in female mice. Cardiovasc Diabetol 16: 61, 2017. doi:10.1186/s12933-017-0544-4. - DOI - PMC - PubMed
-
- Babaei S, Picard P, Ravandi A, Monge JC, Lee TC, Cernacek P, Stewart DJ. Blockade of endothelin receptors markedly reduces atherosclerosis in LDL receptor deficient mice: role of endothelin in macrophage foam cell formation. Cardiovasc Res 48: 158–167, 2000. doi:10.1016/S0008-6363(00)00169-3. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous