

Molecular and clinical profile of von Willebrand disease in Spain (PCM-EVW-ES): comprehensive genetic analysis by next-generation sequencing of 480 patients
- PMID: 28971901
- PMCID: PMC5709099
- DOI: 10.3324/haematol.2017.168765
Molecular and clinical profile of von Willebrand disease in Spain (PCM-EVW-ES): comprehensive genetic analysis by next-generation sequencing of 480 patients
Abstract
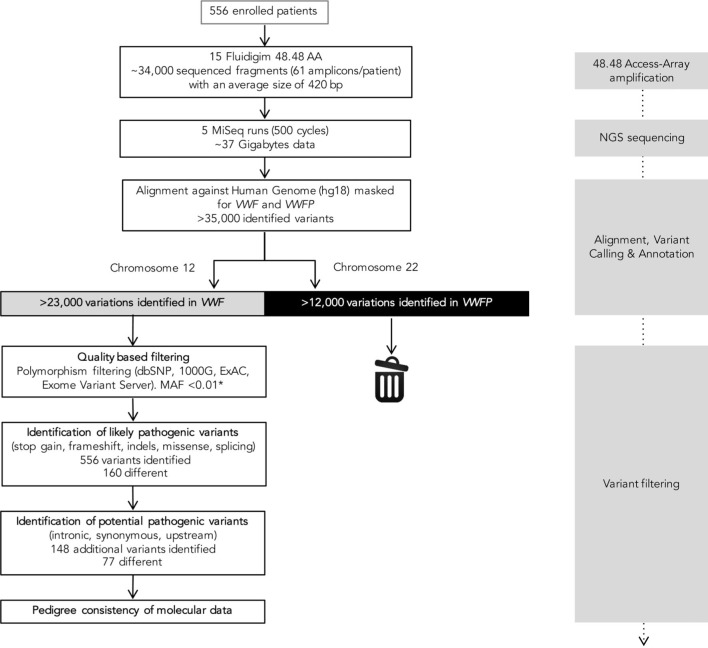
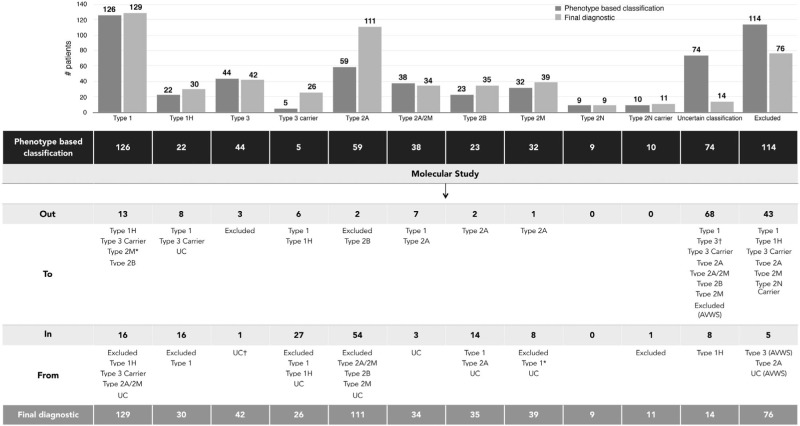
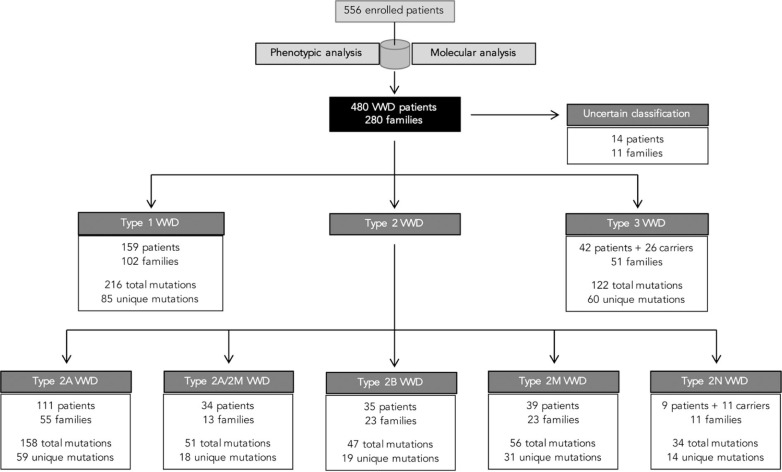
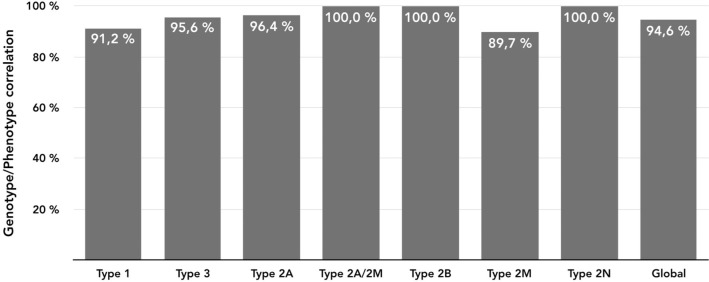
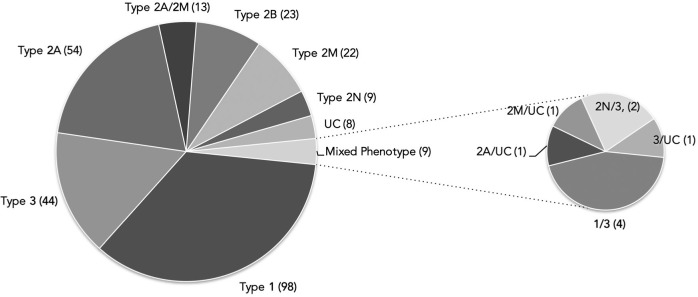
Molecular diagnosis of patients with von Willebrand disease is pending in most populations due to the complexity and high cost of conventional molecular analyses. The need for molecular and clinical characterization of von Willebrand disease in Spain prompted the creation of a multicenter project (PCM-EVW-ES) that resulted in the largest prospective cohort study of patients with all types of von Willebrand disease. Molecular analysis of relevant regions of the VWF, including intronic and promoter regions, was achieved in the 556 individuals recruited via the development of a simple, innovative, relatively low-cost protocol based on microfluidic technology and next-generation sequencing. A total of 704 variants (237 different) were identified along VWF, 155 of which had not been previously recorded in the international mutation database. The potential pathogenic effect of these variants was assessed by in silico analysis. Furthermore, four short tandem repeats were analyzed in order to evaluate the ancestral origin of recurrent mutations. The outcome of genetic analysis allowed for the reclassification of 110 patients, identification of 37 asymptomatic carriers (important for genetic counseling) and re-inclusion of 43 patients previously excluded by phenotyping results. In total, 480 patients were definitively diagnosed. Candidate mutations were identified in all patients except 13 type 1 von Willebrand disease, yielding a high genotype-phenotype correlation. Our data reinforce the capital importance and usefulness of genetics in von Willebrand disease diagnostics. The progressive implementation of molecular study as the first-line test for routine diagnosis of this condition will lead to increasingly more personalized and effective care for this patient population.
Copyright© 2017 Ferrata Storti Foundation.
Figures
References
-
- Sadler JE, Budde U, Eikenboom JC, et al. Update on the pathophysiology and classification of von Willebrand disease: a report of the Subcommittee on von Willebrand Factor. J Thromb Haemost. 2006;4(10): 2103–2114. - PubMed
-
- Rodeghiero F, Castaman G, Dini E. Epidemiological investigation of the prevalence of von Willebrand’s disease. Blood. 1987;69(2):454–459. - PubMed
-
- Ruggeri ZM. Von Willebrand factor, platelets and endothelial cell interactions. J Thromb Haemost. 2003;1(7):13351–1342. - PubMed
-
- Vlot AJ, Koppelman SJ, Meijers JC, et al. Kinetics of factor VIII-von Willebrand factor association. Blood. 1996;87(5):1809–1816. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
