

Activation of the sweet taste receptor, T1R3, by the artificial sweetener sucralose regulates the pulmonary endothelium
- PMID: 28971978
- PMCID: PMC5866431
- DOI: 10.1152/ajplung.00490.2016
Activation of the sweet taste receptor, T1R3, by the artificial sweetener sucralose regulates the pulmonary endothelium
Abstract
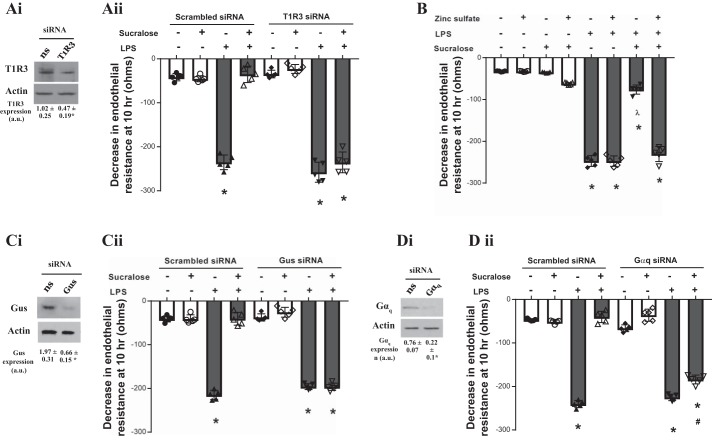
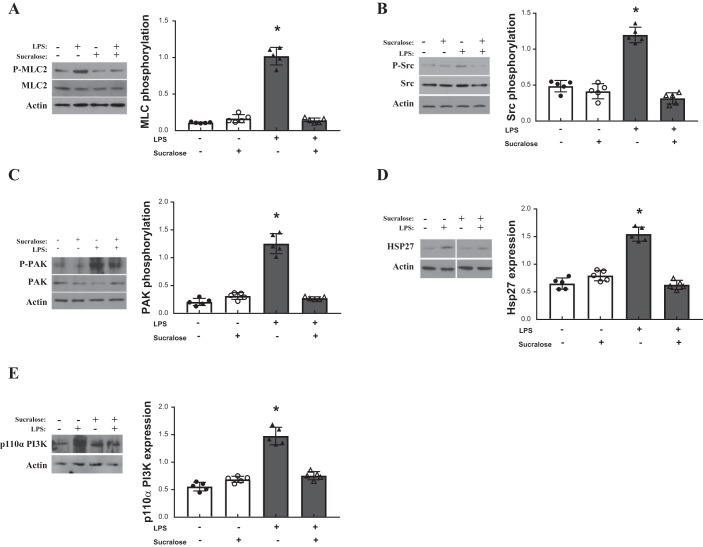
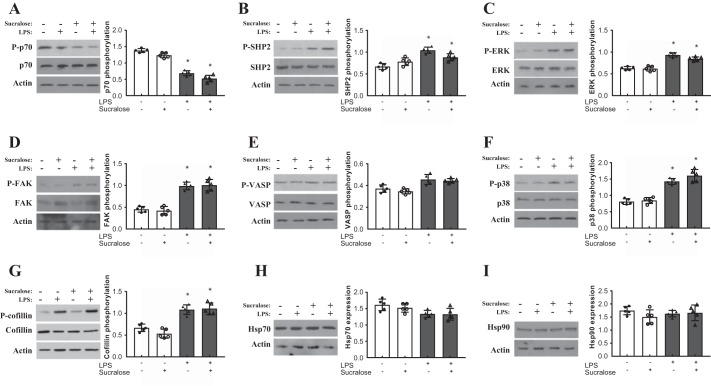
A hallmark of acute respiratory distress syndrome (ARDS) is pulmonary vascular permeability. In these settings, loss of barrier integrity is mediated by cell-contact disassembly and actin remodeling. Studies into molecular mechanisms responsible for improving microvascular barrier function are therefore vital in the development of therapeutic targets for reducing vascular permeability in ARDS. The sweet taste receptor T1R3 is a G protein-coupled receptor, activated following exposure to sweet molecules, to trigger a gustducin-dependent signal cascade. In recent years, extraoral locations for T1R3 have been identified; however, no studies have focused on T1R3 within the vasculature. We hypothesize that activation of T1R3, in the pulmonary vasculature, plays a role in regulating endothelial barrier function in settings of ARDS. Our study demonstrated expression of T1R3 within the pulmonary vasculature, with a drop in expression levels following exposure to barrier-disruptive agents. Exposure of lung microvascular endothelial cells to the intensely sweet molecule sucralose attenuated LPS- and thrombin-induced endothelial barrier dysfunction. Likewise, sucralose exposure attenuated bacteria-induced lung edema formation in vivo. Inhibition of sweet taste signaling, through zinc sulfate, T1R3, or G-protein siRNA, blunted the protective effects of sucralose on the endothelium. Sucralose significantly reduced LPS-induced increased expression or phosphorylation of the key signaling molecules Src, p21-activated kinase (PAK), myosin light chain-2 (MLC2), heat shock protein 27 (HSP27), and p110α phosphatidylinositol 3-kinase (p110αPI3K). Activation of T1R3 by sucralose protects the pulmonary endothelium from edemagenic agent-induced barrier disruption, potentially through abrogation of Src/PAK/p110αPI3K-mediated cell-contact disassembly and Src/MLC2/HSP27-mediated actin remodeling. Identification of sweet taste sensing in the pulmonary vasculature may represent a novel therapeutic target to protect the endothelium in settings of ARDS.
Keywords: T1R3; acute respiratory distress syndrome; artificial sweeteners; pulmonary endothelium; sweet taste.
Figures
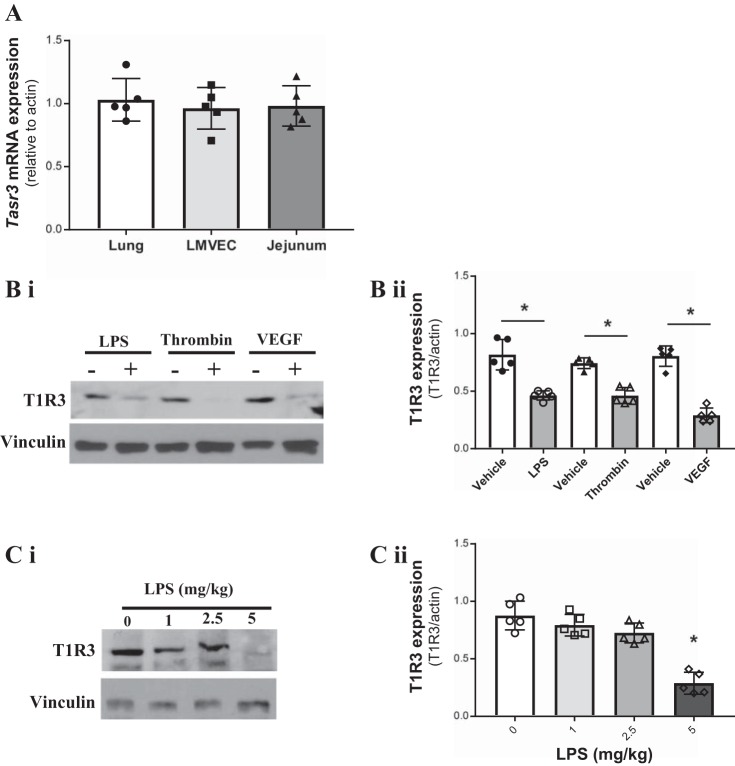
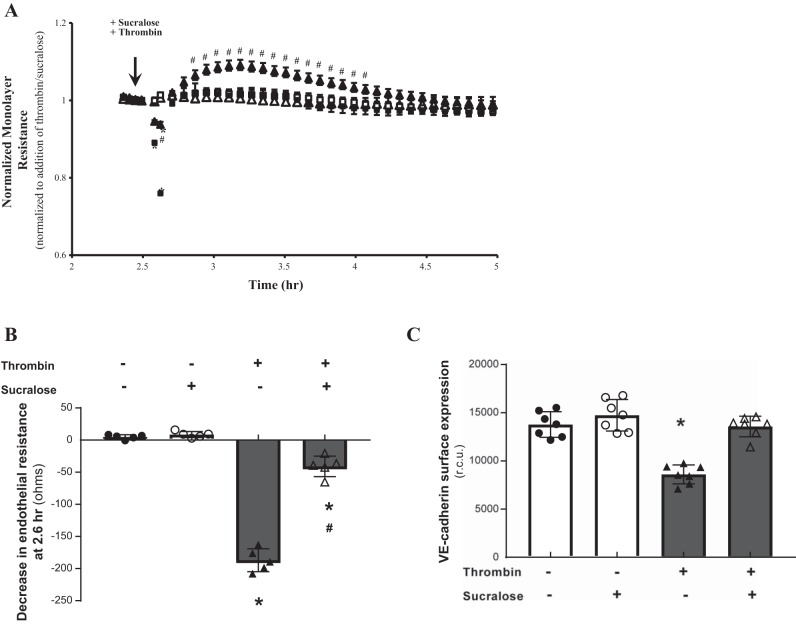
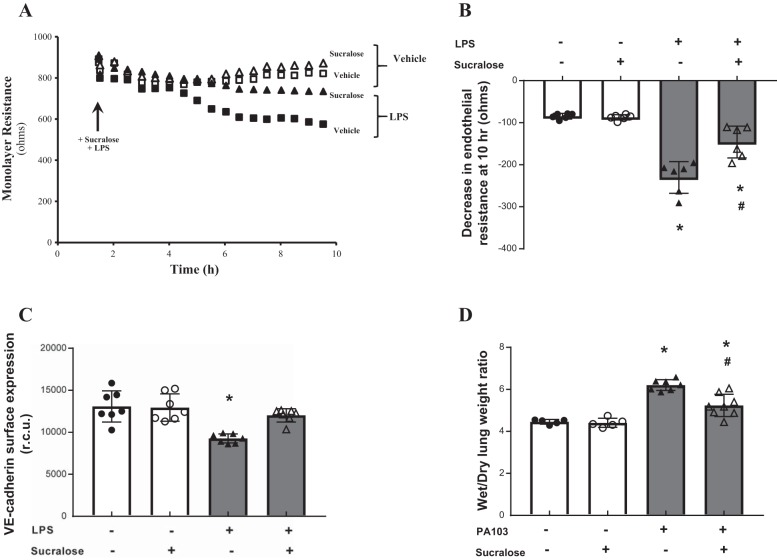
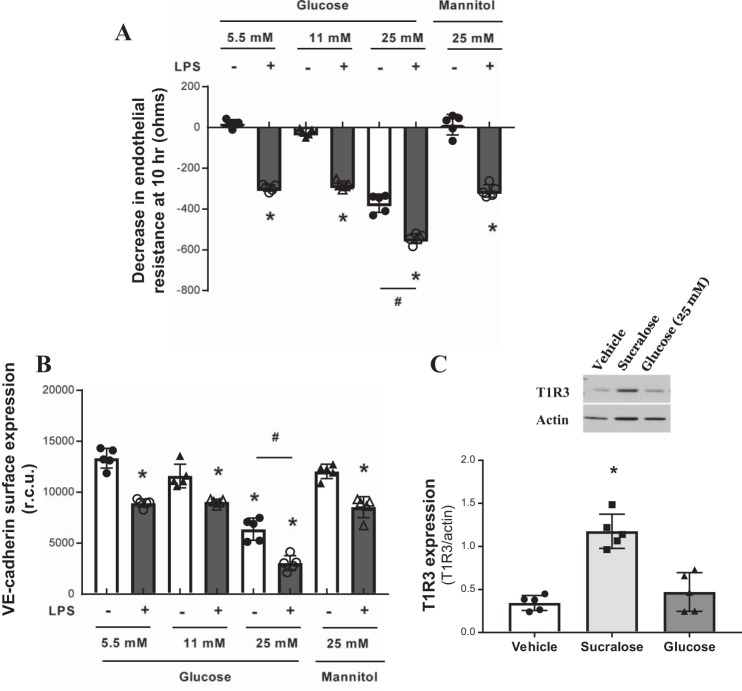
References
-
- Alvarado AG, Thiagarajan PS, Mulkearns-Hubert EE, Silver DJ, Hale JS, Alban TJ, Turaga SM, Jarrar A, Reizes O, Longworth MS, Vogelbaum MA, Lathia JD. Glioblastoma cancer stem cells evade innate immune suppression of self-renewal through reduced TLR4 expression. Cell Stem Cell 20: 450–461.e4, 2017. doi: 10.1016/j.stem.2016.12.001. - DOI - PMC - PubMed
-
- Aoki T, Tsunekawa K, Araki O, Ogiwara T, Nara M, Sumino H, Kimura T, Murakami M. Type 2 iodothyronine deiodinase activity is required for rapid stimulation of PI3K by thyroxine in human umbilical vein endothelial cells. Endocrinology 156: 4312–4324, 2015. doi: 10.1210/en.2014-1988. - DOI - PMC - PubMed
-
- Barabutis N, Handa V, Dimitropoulou C, Rafikov R, Snead C, Kumar S, Joshi A, Thangjam G, Fulton D, Black SM, Patel V, Catravas JD. LPS induces pp60c-src-mediated tyrosine phosphorylation of Hsp90 in lung vascular endothelial cells and mouse lung. Am J Physiol Lung Cell Mol Physiol 304: L883–L893, 2013. doi: 10.1152/ajplung.00419.2012. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous