

Phenotypic Characterization of EIF2AK4 Mutation Carriers in a Large Cohort of Patients Diagnosed Clinically With Pulmonary Arterial Hypertension
- PMID: 28972005
- PMCID: PMC5700414
- DOI: 10.1161/CIRCULATIONAHA.117.028351
Phenotypic Characterization of EIF2AK4 Mutation Carriers in a Large Cohort of Patients Diagnosed Clinically With Pulmonary Arterial Hypertension
Abstract
Background: Pulmonary arterial hypertension (PAH) is a rare disease with an emerging genetic basis. Heterozygous mutations in the gene encoding the bone morphogenetic protein receptor type 2 (BMPR2) are the commonest genetic cause of PAH, whereas biallelic mutations in the eukaryotic translation initiation factor 2 alpha kinase 4 gene (EIF2AK4) are described in pulmonary veno-occlusive disease/pulmonary capillary hemangiomatosis. Here, we determine the frequency of these mutations and define the genotype-phenotype characteristics in a large cohort of patients diagnosed clinically with PAH.
Methods: Whole-genome sequencing was performed on DNA from patients with idiopathic and heritable PAH and with pulmonary veno-occlusive disease/pulmonary capillary hemangiomatosis recruited to the National Institute of Health Research BioResource-Rare Diseases study. Heterozygous variants in BMPR2 and biallelic EIF2AK4 variants with a minor allele frequency of <1:10 000 in control data sets and predicted to be deleterious (by combined annotation-dependent depletion, PolyPhen-2, and sorting intolerant from tolerant predictions) were identified as potentially causal. Phenotype data from the time of diagnosis were also captured.
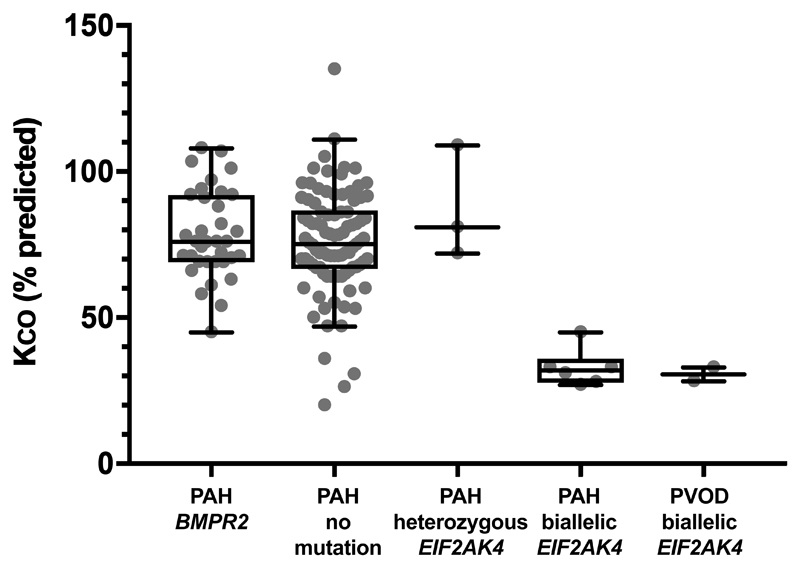
Results: Eight hundred sixty-four patients with idiopathic or heritable PAH and 16 with pulmonary veno-occlusive disease/pulmonary capillary hemangiomatosis were recruited. Mutations in BMPR2 were identified in 130 patients (14.8%). Biallelic mutations in EIF2AK4 were identified in 5 patients with a clinical diagnosis of pulmonary veno-occlusive disease/pulmonary capillary hemangiomatosis. Furthermore, 9 patients with a clinical diagnosis of PAH carried biallelic EIF2AK4 mutations. These patients had a reduced transfer coefficient for carbon monoxide (Kco; 33% [interquartile range, 30%-35%] predicted) and younger age at diagnosis (29 years; interquartile range, 23-38 years) and more interlobular septal thickening and mediastinal lymphadenopathy on computed tomography of the chest compared with patients with PAH without EIF2AK4 mutations. However, radiological assessment alone could not accurately identify biallelic EIF2AK4 mutation carriers. Patients with PAH with biallelic EIF2AK4 mutations had a shorter survival.
Conclusions: Biallelic EIF2AK4 mutations are found in patients classified clinically as having idiopathic and heritable PAH. These patients cannot be identified reliably by computed tomography, but a low Kco and a young age at diagnosis suggests the underlying molecular diagnosis. Genetic testing can identify these misclassified patients, allowing appropriate management and early referral for lung transplantation.
Keywords: genetics; hypertension, pulmonary; mutation; prognosis; pulmonary veno-occlusive disease.
© 2017 The Authors.
Figures
Comment in
-
Pulmonary Veno-Occlusive Disease: Welcome to the PAHty (Bostonian for Party).Circulation. 2017 Nov 21;136(21):2034-2036. doi: 10.1161/CIRCULATIONAHA.117.031158. Circulation. 2017. PMID: 29158214 No abstract available.
-
Letter by Hernandez-Gonzalez et al Regarding Article, "Phenotypic Characterization of EIF2AK4 Mutation Carriers in a Large Cohort of Patients Diagnosed Clinically With Pulmonary Arterial Hypertension".Circulation. 2018 May 29;137(22):2411-2412. doi: 10.1161/CIRCULATIONAHA.118.033641. Circulation. 2018. PMID: 29844074 No abstract available.
-
Response by Hadinnapola et al to Letter Regarding Article, "Phenotypic Characterization of EIF2AK4 Mutation Carriers in a Large Cohort of Patients Diagnosed Clinically With Pulmonary Arterial Hypertension".Circulation. 2018 May 29;137(22):2413-2414. doi: 10.1161/CIRCULATIONAHA.118.033970. Circulation. 2018. PMID: 29844075 No abstract available.
Similar articles
-
EIF2AK4 mutation as "second hit" in hereditary pulmonary arterial hypertension.Respir Res. 2016 Nov 4;17(1):141. doi: 10.1186/s12931-016-0457-x. Respir Res. 2016. PMID: 27809840 Free PMC article.
-
Clinical characteristics and survival of Chinese patients diagnosed with pulmonary arterial hypertension who carry BMPR2 or EIF2KAK4 variants.BMC Pulm Med. 2020 May 29;20(1):150. doi: 10.1186/s12890-020-01179-7. BMC Pulm Med. 2020. PMID: 32471403 Free PMC article.
-
EIF2AK4 Mutations in Patients Diagnosed With Pulmonary Arterial Hypertension.Chest. 2017 Apr;151(4):821-828. doi: 10.1016/j.chest.2016.11.014. Epub 2016 Nov 22. Chest. 2017. PMID: 27884767
-
Genetics of pulmonary hypertension in the clinic.Curr Opin Pulm Med. 2017 Sep;23(5):386-391. doi: 10.1097/MCP.0000000000000414. Curr Opin Pulm Med. 2017. PMID: 28661905 Review.
-
Heritable pulmonary hypertension: from bench to bedside.Eur Respir Rev. 2017 Sep 6;26(145):170037. doi: 10.1183/16000617.0037-2017. Print 2017 Sep 30. Eur Respir Rev. 2017. PMID: 28877973 Free PMC article. Review.
Cited by
-
Survival outcomes in EIF2AK4 mutation-associated pulmonary arterial hypertension: seeking clarity in contrast.Eur Heart J Case Rep. 2024 Oct 7;8(11):ytae538. doi: 10.1093/ehjcr/ytae538. eCollection 2024 Nov. Eur Heart J Case Rep. 2024. PMID: 39502262 Free PMC article.
-
Pulmonary Arterial Hypertension with Features of Venous Involvement: A Detective's Task.Arq Bras Cardiol. 2024 Apr 29;121(4):e20230565. doi: 10.36660/abc.20230565. eCollection 2024. Arq Bras Cardiol. 2024. PMID: 38695472 Free PMC article. English, Portuguese.
-
An interesting case of progressive dyspnoea and diffuse mediastinal adenopathy in a 25-year-old man.Breathe (Sheff). 2021 Mar;17(1):200289. doi: 10.1183/20734735.0289-2020. Breathe (Sheff). 2021. PMID: 34295399 Free PMC article.
-
The role of genomics and genetics in pulmonary arterial hypertension.Glob Cardiol Sci Pract. 2020 Apr 30;2020(1):e202013. doi: 10.21542/gcsp.2020.13. Glob Cardiol Sci Pract. 2020. PMID: 33150157 Free PMC article. Review. No abstract available.
-
Precision Medicine for Pulmonary Vascular Disease: The Future Is Now (2023 Grover Conference Series).Pulm Circ. 2025 Jan 2;15(1):e70027. doi: 10.1002/pul2.70027. eCollection 2025 Jan. Pulm Circ. 2025. PMID: 39749110 Free PMC article.
References
-
- Simonneau G, Gatzoulis MA, Adatia I, Celermajer D, Denton C, Ghofrani A, Gomez Sanchez MA, Krishna Kumar R, Landzberg M, Machado RF, Olschewski H, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2013;62:D34–41. - PubMed
-
- Galie N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, Simonneau G, Peacock A, Vonk Noordegraaf A, Beghetti M, Ghofrani A, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT) Eur Respir J. 2015;46:903–75. - PubMed
-
- Resten A, Maitre S, Humbert M, Rabiller A, Sitbon O, Capron F, Simonneau G, Musset D. Pulmonary hypertension: CT of the chest in pulmonary venoocclusive disease. AJR Am J Roentgenol. 2004;183:65–70. - PubMed
-
- Montani D, Achouh L, Dorfmuller P, Le Pavec J, Sztrymf B, Tcherakian C, Rabiller A, Haque R, Sitbon O, Jais X, Dartevelle P, et al. Pulmonary veno-occlusive disease: clinical, functional, radiologic, and hemodynamic characteristics and outcome of 24 cases confirmed by histology. Medicine (Baltimore) 2008;87:220–33. - PubMed
-
- Trip P, Girerd B, Bogaard HJ, de Man FS, Boonstra A, Garcia G, Humbert M, Montani D, Vonk-Noordegraaf A. Diffusion capacity and BMPR2 mutations in pulmonary arterial hypertension. Eur Respir J. 2014;43:1195–8. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
