

Pharmacochaperoning in a Drosophila model system rescues human dopamine transporter variants associated with infantile/juvenile parkinsonism
- PMID: 28972153
- PMCID: PMC5702666
- DOI: 10.1074/jbc.M117.797092
Pharmacochaperoning in a Drosophila model system rescues human dopamine transporter variants associated with infantile/juvenile parkinsonism
Abstract
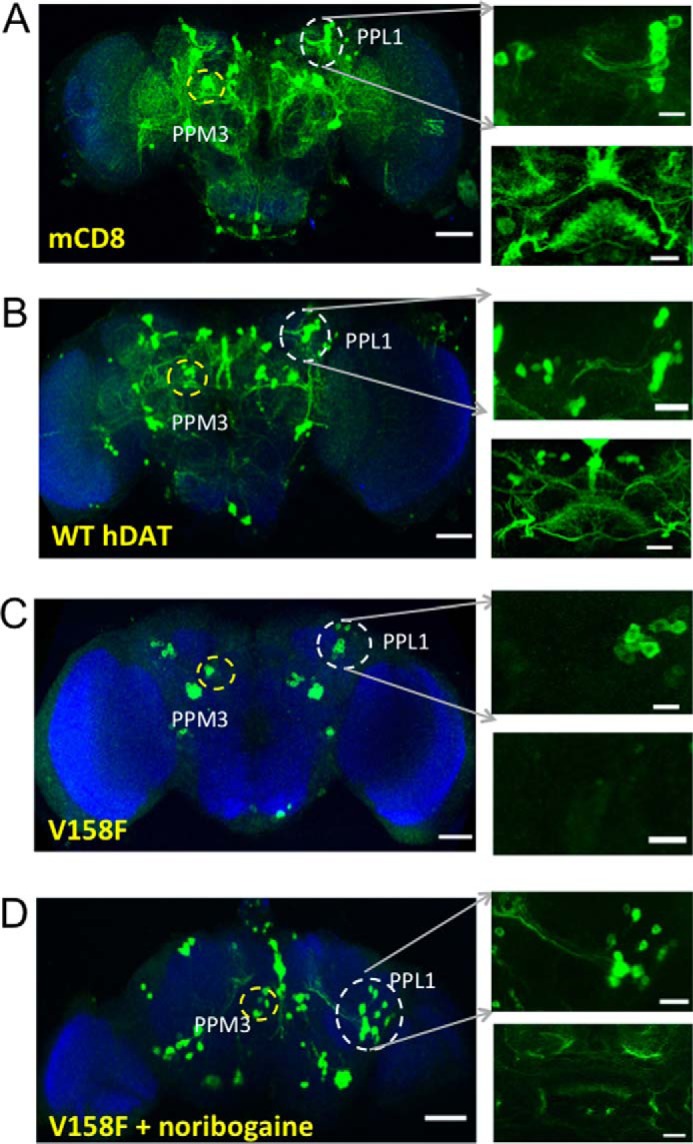
Point mutations in the gene encoding the human dopamine transporter (hDAT, SLC6A3) cause a syndrome of infantile/juvenile dystonia and parkinsonism. To unravel the molecular mechanism underlying these disorders and investigate possible pharmacological therapies, here we examined 13 disease-causing DAT mutants that were retained in the endoplasmic reticulum when heterologously expressed in HEK293 cells. In three of these mutants, i.e. hDAT-V158F, hDAT-G327R, and hDAT-L368Q, the folding deficit was remedied with the pharmacochaperone noribogaine or the heat shock protein 70 (HSP70) inhibitor pifithrin-μ such that endoplasmic reticulum export of and radioligand binding and substrate uptake by these DAT mutants were restored. In Drosophila melanogaster, DAT deficiency results in reduced sleep. We therefore exploited the power of targeted transgene expression of mutant hDAT in Drosophila to explore whether these hDAT mutants could also be pharmacologically rescued in an intact organism. Noribogaine or pifithrin-μ treatment supported hDAT delivery to the presynaptic terminals of dopaminergic neurons and restored sleep to normal length in DAT-deficient (fumin) Drosophila lines expressing hDAT-V158F or hDAT-G327R. In contrast, expression of hDAT-L368Q in the Drosophila DAT mutant background caused developmental lethality, indicating a toxic action not remedied by pharmacochaperoning. Our observations identified those mutations most likely amenable to pharmacological rescue in the affected children. In addition, our findings also highlight the challenges of translating insights from pharmacochaperoning in cell culture to the clinical situation. Because of the evolutionary conservation in dopaminergic neurotransmission between Drosophila and people, pharmacochaperoning of DAT in D. melanogaster may allow us to bridge that gap.
Keywords: chaperone; dopamine; dopamine transporter; endoplasmic reticulum (ER); neurotransmitter transport.
© 2017 by The American Society for Biochemistry and Molecular Biology, Inc.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures










References
-
- Carta E., Chung S. K., James V. M., Robinson A., Gill J. L., Remy N., Vanbellinghen J. F., Drew C. J., Cagdas S., Cameron D., Cowan F. M., Del Toro M., Graham G. E., Manzur A. Y., Masri A., et al. (2012) Mutations in the GlyT2 gene (SLC6A5) are a second major cause of startle disease. J. Biol. Chem. 287, 28975–28985 - PMC - PubMed
-
- Kurian M. A., Zhen J., Cheng S. Y., Li Y., Mordekar S. R., Jardine P., Morgan N. V., Meyer E., Tee L., Pasha S., Wassmer E., Heales S. J., Gissen P., Reith M. E., and Maher E. R. (2009) Homozygous loss-of-function mutations in the gene encoding the dopamine transporter are associated with infantile parkinsonism-dystonia. J. Clin. Invest. 119, 1595–1603 - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
