

Mutual synergy between catalase and peroxidase activities of the bifunctional enzyme KatG is facilitated by electron hole-hopping within the enzyme
- PMID: 28972181
- PMCID: PMC5682954
- DOI: 10.1074/jbc.M117.791202
Mutual synergy between catalase and peroxidase activities of the bifunctional enzyme KatG is facilitated by electron hole-hopping within the enzyme
Abstract
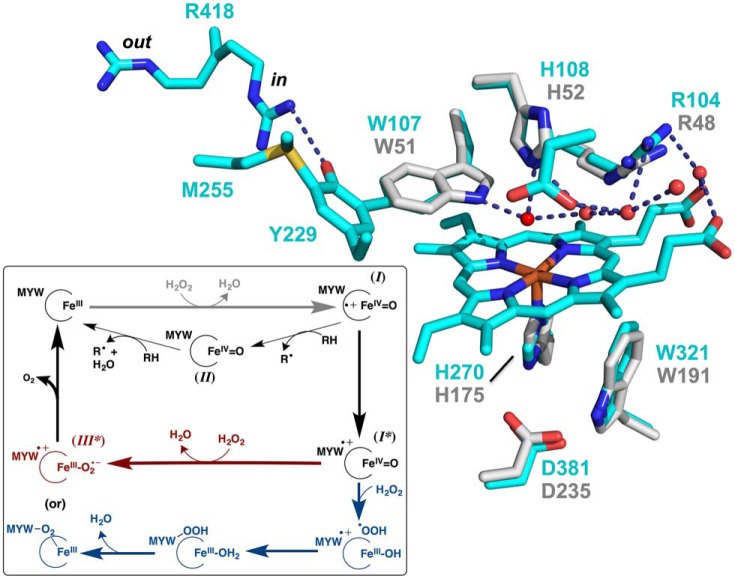
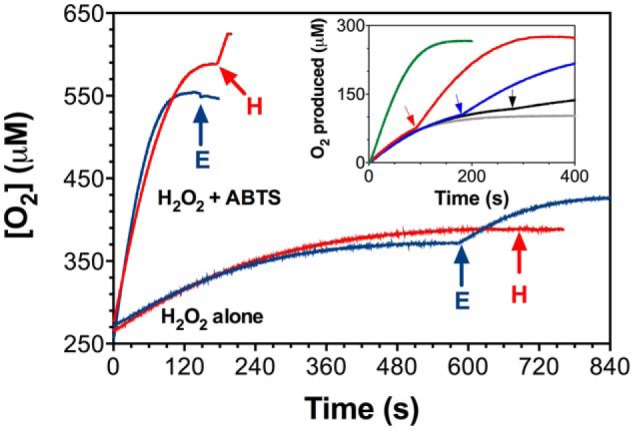
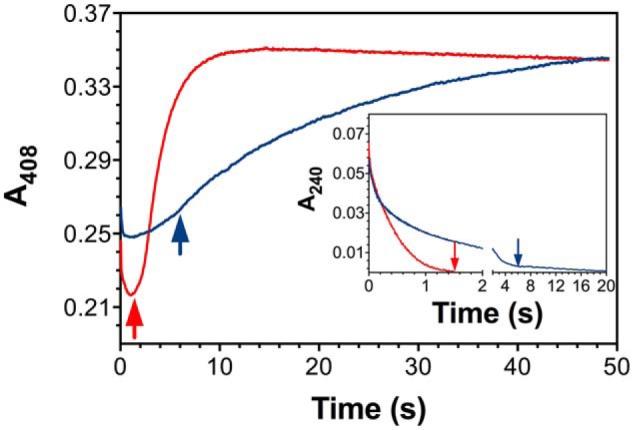
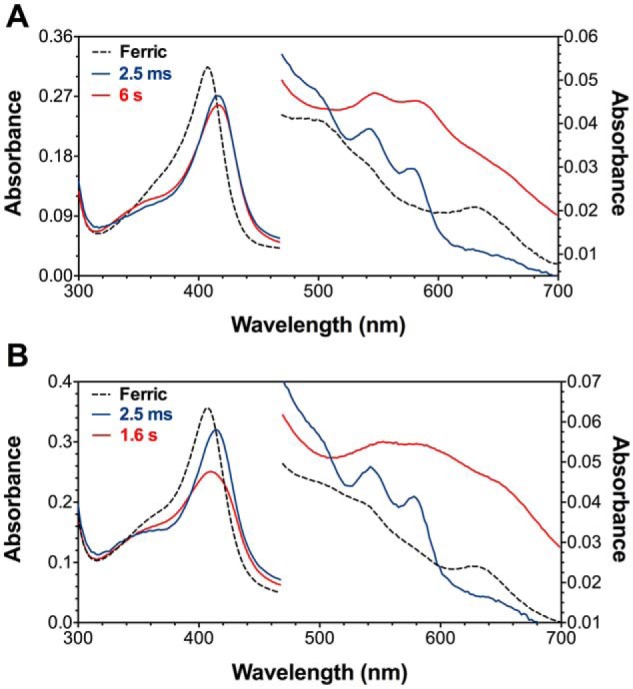
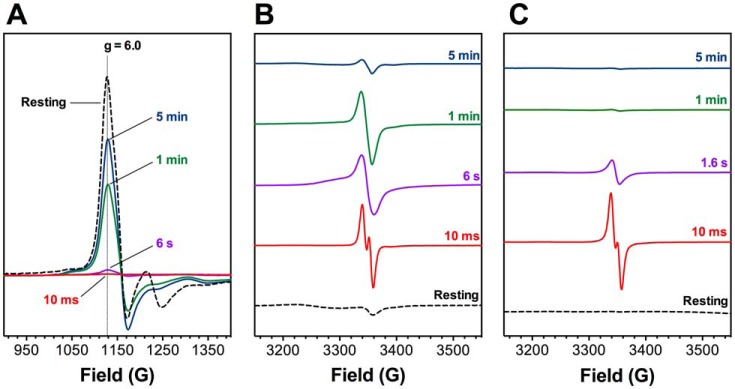
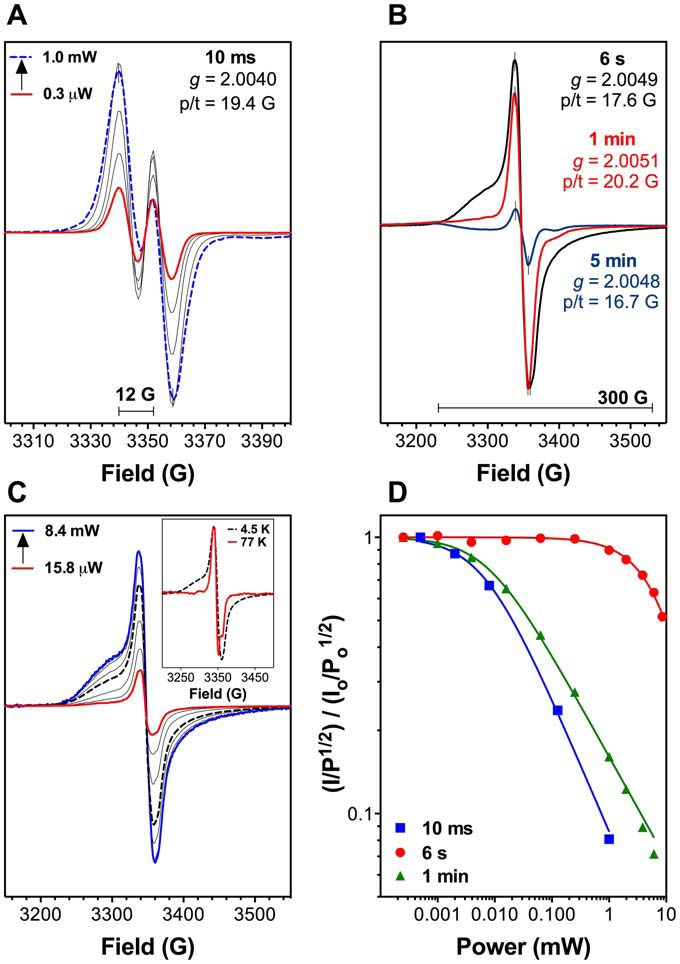
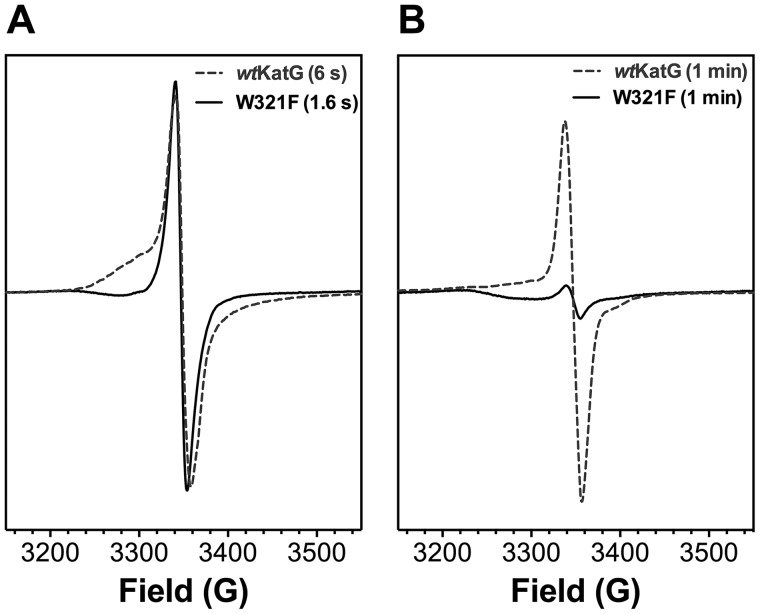
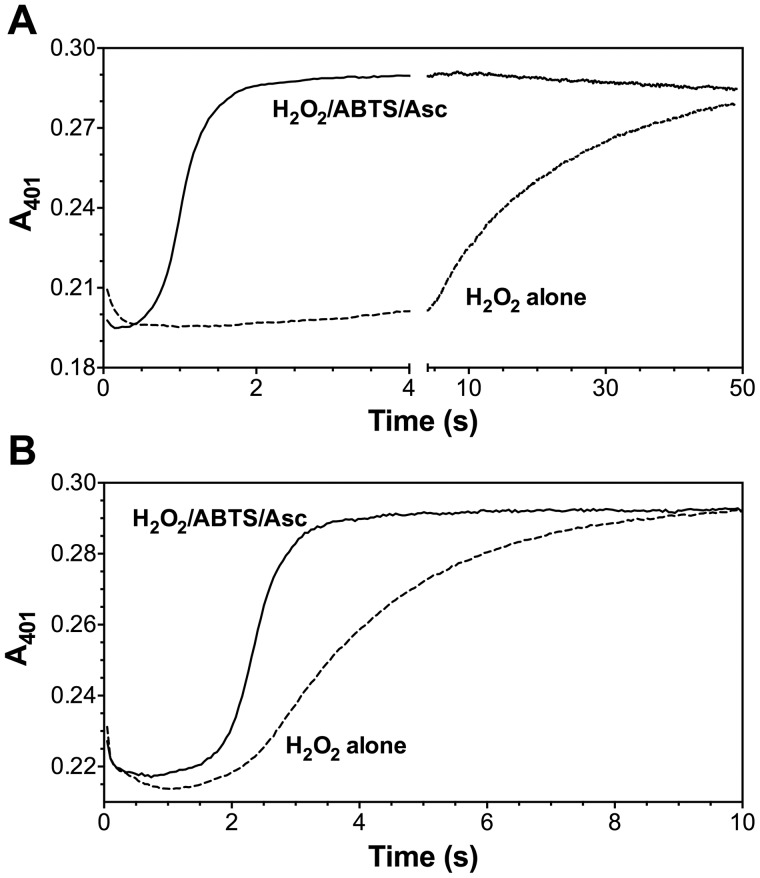
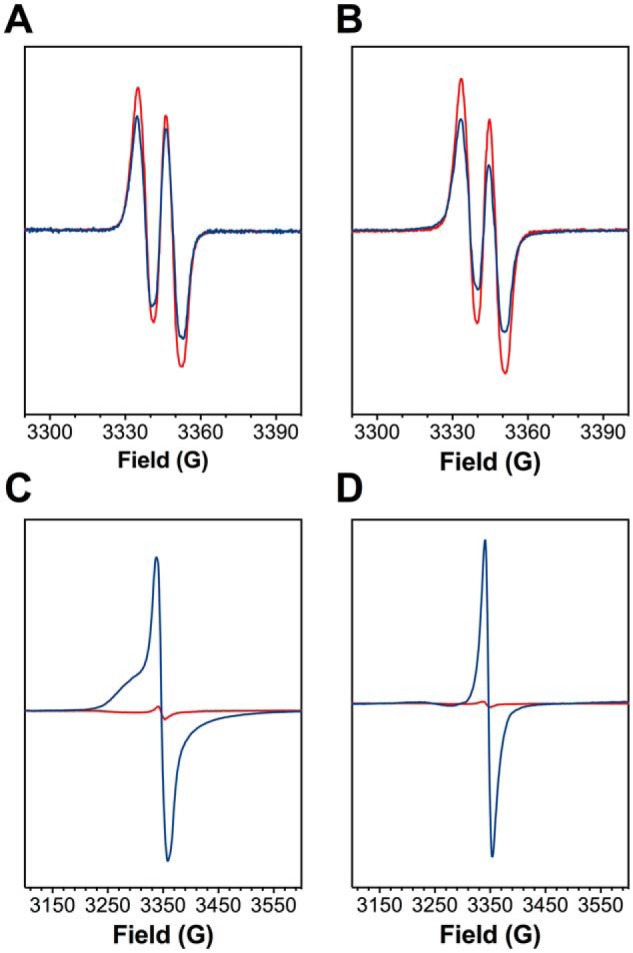
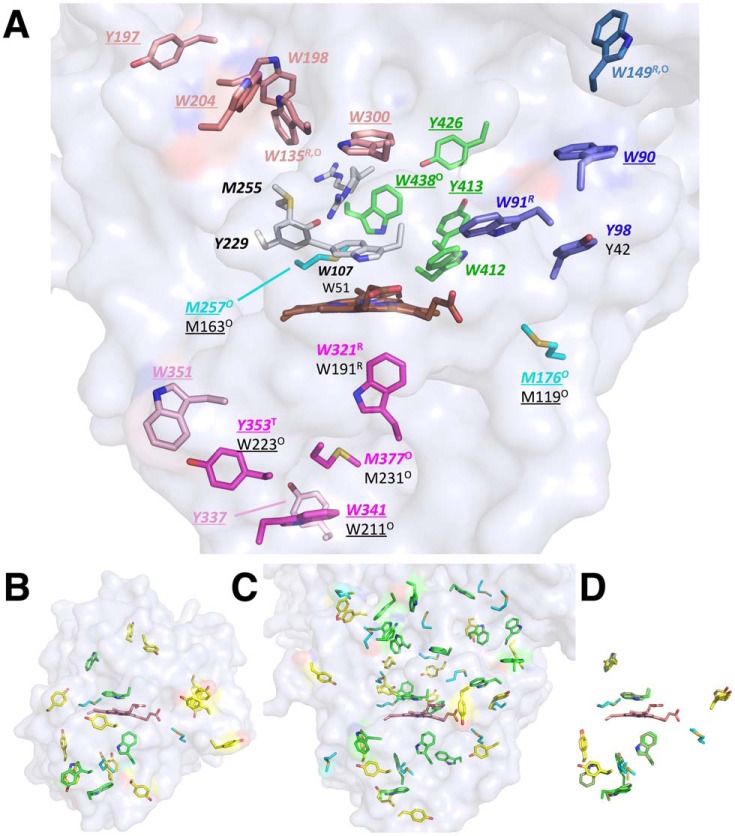
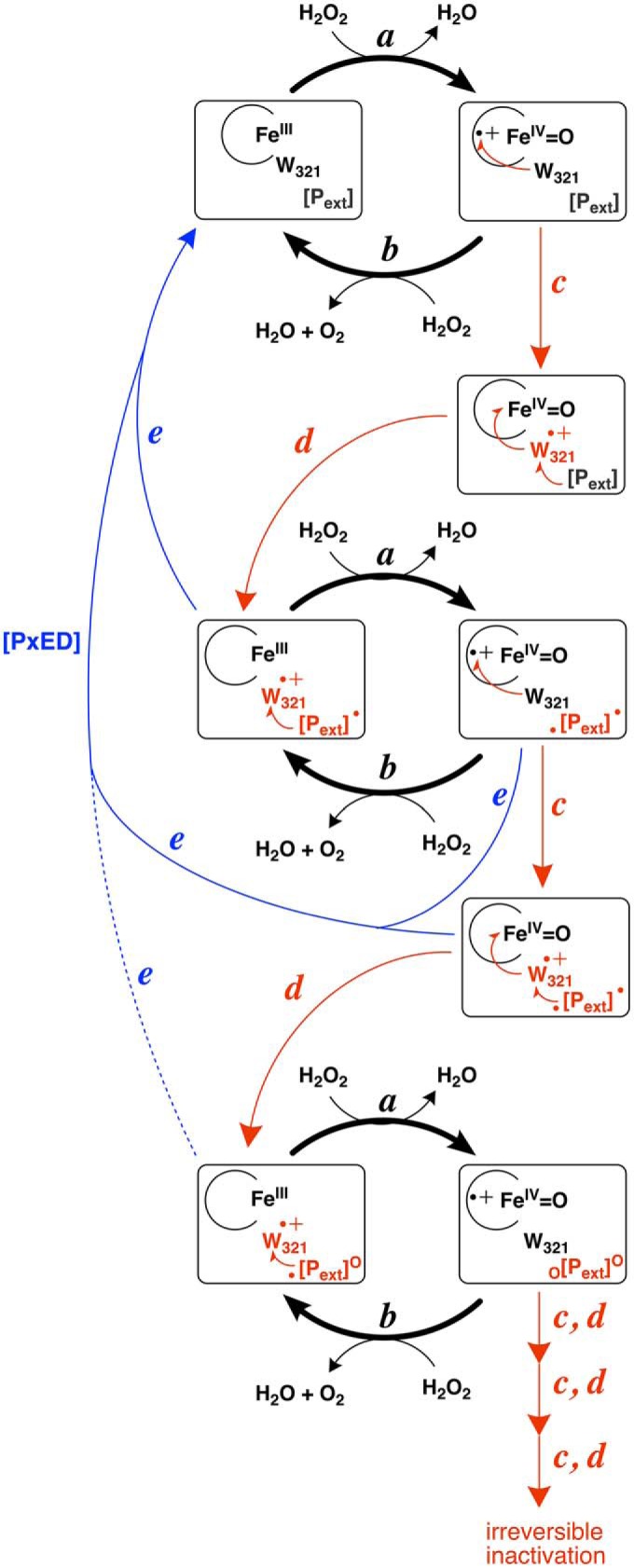
KatG is a bifunctional, heme-dependent enzyme in the front-line defense of numerous bacterial and fungal pathogens against H2O2-induced oxidative damage from host immune responses. Contrary to the expectation that catalase and peroxidase activities should be mutually antagonistic, peroxidatic electron donors (PxEDs) enhance KatG catalase activity. Here, we establish the mechanism of synergistic cooperation between these activities. We show that at low pH values KatG can fully convert H2O2 to O2 and H2O only if a PxED is present in the reaction mixture. Stopped-flow spectroscopy results indicated rapid initial rates of H2O2 disproportionation slowing concomitantly with the accumulation of ferryl-like heme states. These states very slowly returned to resting (i.e. ferric) enzyme, indicating that they represented catalase-inactive intermediates. We also show that an active-site tryptophan, Trp-321, participates in off-pathway electron transfer. A W321F variant in which the proximal tryptophan was replaced with a non-oxidizable phenylalanine exhibited higher catalase activity and less accumulation of off-pathway heme intermediates. Finally, rapid freeze-quench EPR experiments indicated that both WT and W321F KatG produce the same methionine-tyrosine-tryptophan (MYW) cofactor radical intermediate at the earliest reaction time points and that Trp-321 is the preferred site of off-catalase protein oxidation in the native enzyme. Of note, PxEDs did not affect the formation of the MYW cofactor radical but could reduce non-productive protein-based radical species that accumulate during reaction with H2O2 Our results suggest that catalase-inactive intermediates accumulate because of off-mechanism oxidation, primarily of Trp-321, and PxEDs stimulate KatG catalase activity by preventing the accumulation of inactive intermediates.
Keywords: electron paramagnetic resonance (EPR); electron transfer; enzyme inactivation; free radicals; heme; hydrogen peroxide; tryptophan.
© 2017 by The American Society for Biochemistry and Molecular Biology, Inc.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures
Similar articles
-
An oxyferrous heme/protein-based radical intermediate is catalytically competent in the catalase reaction of Mycobacterium tuberculosis catalase-peroxidase (KatG).J Biol Chem. 2009 Mar 13;284(11):7017-29. doi: 10.1074/jbc.M808106200. Epub 2009 Jan 12. J Biol Chem. 2009. PMID: 19139099 Free PMC article.
-
Specific function of the Met-Tyr-Trp adduct radical and residues Arg-418 and Asp-137 in the atypical catalase reaction of catalase-peroxidase KatG.J Biol Chem. 2012 Oct 26;287(44):37057-65. doi: 10.1074/jbc.M112.401208. Epub 2012 Aug 23. J Biol Chem. 2012. PMID: 22918833 Free PMC article.
-
Catalase in peroxidase clothing: Interdependent cooperation of two cofactors in the catalytic versatility of KatG.Arch Biochem Biophys. 2014 Feb 15;544:27-39. doi: 10.1016/j.abb.2013.11.007. Epub 2013 Nov 23. Arch Biochem Biophys. 2014. PMID: 24280274 Review.
-
Two [Fe(IV)=O Trp*] intermediates in M. tuberculosis catalase-peroxidase discriminated by multifrequency (9-285 GHz) EPR spectroscopy: reactivity toward isoniazid.J Am Chem Soc. 2007 Dec 26;129(51):15954-63. doi: 10.1021/ja075108u. Epub 2007 Dec 4. J Am Chem Soc. 2007. PMID: 18052167
-
Catalase-peroxidase (KatG): a potential frontier in tuberculosis drug development.Crit Rev Biochem Mol Biol. 2024 Dec;59(6):434-446. doi: 10.1080/10409238.2025.2470630. Epub 2025 Feb 27. Crit Rev Biochem Mol Biol. 2024. PMID: 40013498 Review.
Cited by
-
Indole N-Linked Hydroperoxyl Adduct of Protein-Derived Cofactor Modulating Catalase-Peroxidase Functions.Angew Chem Int Ed Engl. 2024 Dec 2;63(49):e202407018. doi: 10.1002/anie.202407018. Epub 2024 Nov 4. Angew Chem Int Ed Engl. 2024. PMID: 39300819
-
Characterization of a catalase-peroxidase variant (L333V-KatG) identified in an INH-resistant Mycobacterium tuberculosis clinical isolate.Biochem Biophys Rep. 2024 Jan 27;37:101649. doi: 10.1016/j.bbrep.2024.101649. eCollection 2024 Mar. Biochem Biophys Rep. 2024. PMID: 38318524 Free PMC article.
-
Functional and protective hole hopping in metalloenzymes.Chem Sci. 2021 Sep 27;12(42):13988-14003. doi: 10.1039/d1sc04286f. eCollection 2021 Nov 3. Chem Sci. 2021. PMID: 34760183 Free PMC article. Review.
-
A new regime of heme-dependent aromatic oxygenase superfamily.Proc Natl Acad Sci U S A. 2021 Oct 26;118(43):e2106561118. doi: 10.1073/pnas.2106561118. Proc Natl Acad Sci U S A. 2021. PMID: 34667125 Free PMC article.
-
Exploring Therapeutic Potential of Catalase: Strategies in Disease Prevention and Management.Biomolecules. 2024 Jun 14;14(6):697. doi: 10.3390/biom14060697. Biomolecules. 2024. PMID: 38927099 Free PMC article. Review.
References
-
- Zamocky M., and Obinger C. (2010) in Biocatalysis Based on Heme Peroxidases (Ayala M., and Torres E., eds) pp. 7–35, Springer-Verlag, Berlin
-
- Tanabe S., Ishii-Minami N., Saitoh K., Otake Y., Kaku H., Shibuya N., Nishizawa Y., and Minami E. (2011) The role of catalase-peroxidase secreted by Magnaporthe oryzae during early infection of rice cells. Mol. Plant Microbe Interact. 24, 163–171 - PubMed
-
- Uhlich G. A. (2009) KatP contributes to OxyR-regulated hydrogen peroxide resistance in Escherichia coli serotype O157: H7. Microbiology 155, 3589–3598 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
