

Divergent effects of glucose and fructose on hepatic lipogenesis and insulin signaling
- PMID: 28972537
- PMCID: PMC5663363
- DOI: 10.1172/JCI94585
Divergent effects of glucose and fructose on hepatic lipogenesis and insulin signaling
Erratum in
-
Divergent effects of glucose and fructose on hepatic lipogenesis and insulin signaling.J Clin Invest. 2018 Mar 1;128(3):1199. doi: 10.1172/JCI99009. Epub 2018 Mar 1. J Clin Invest. 2018. PMID: 29493547 Free PMC article. No abstract available.
Abstract
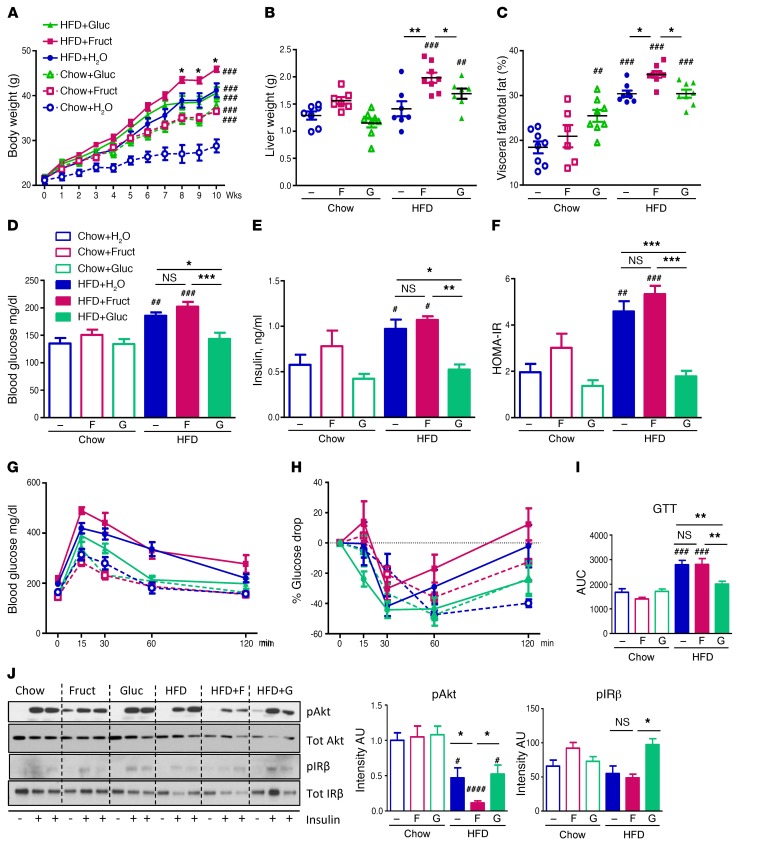
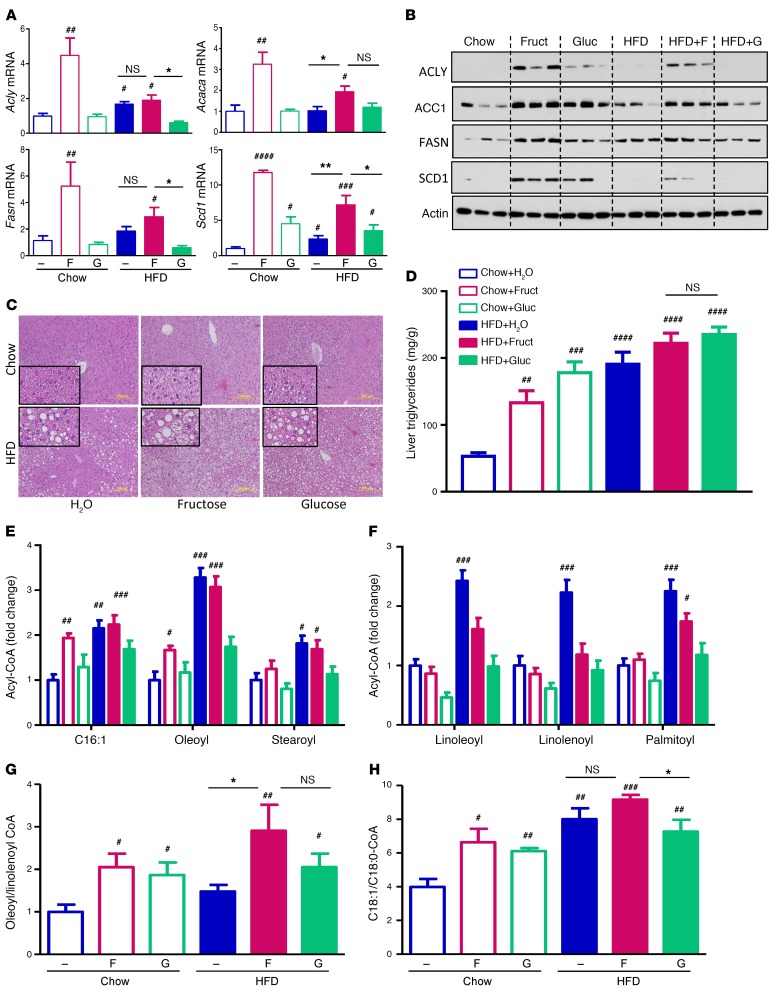
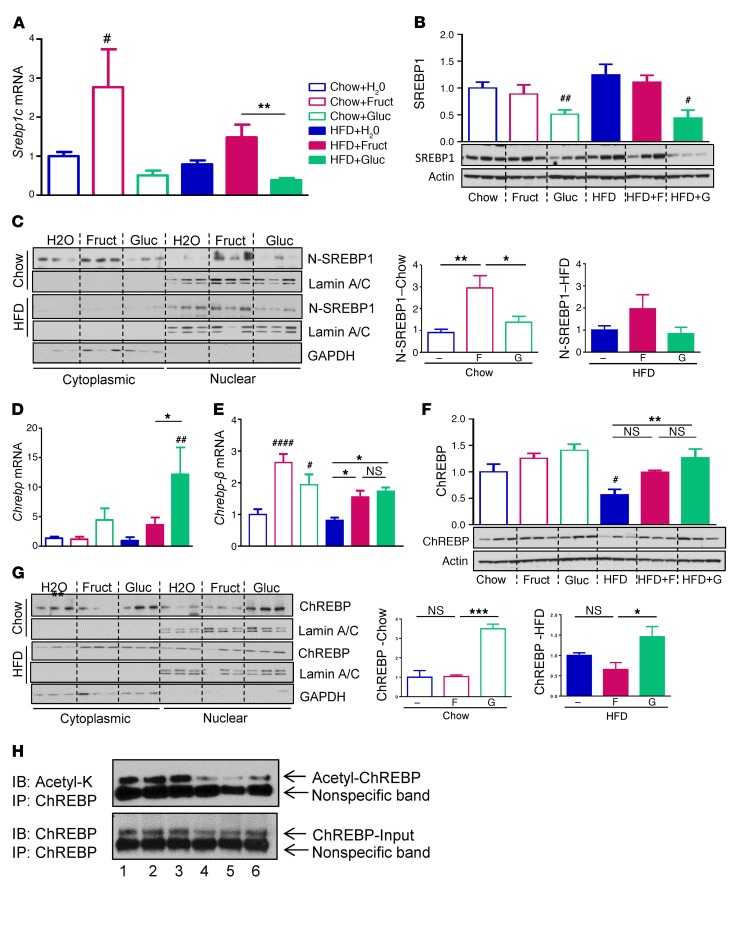
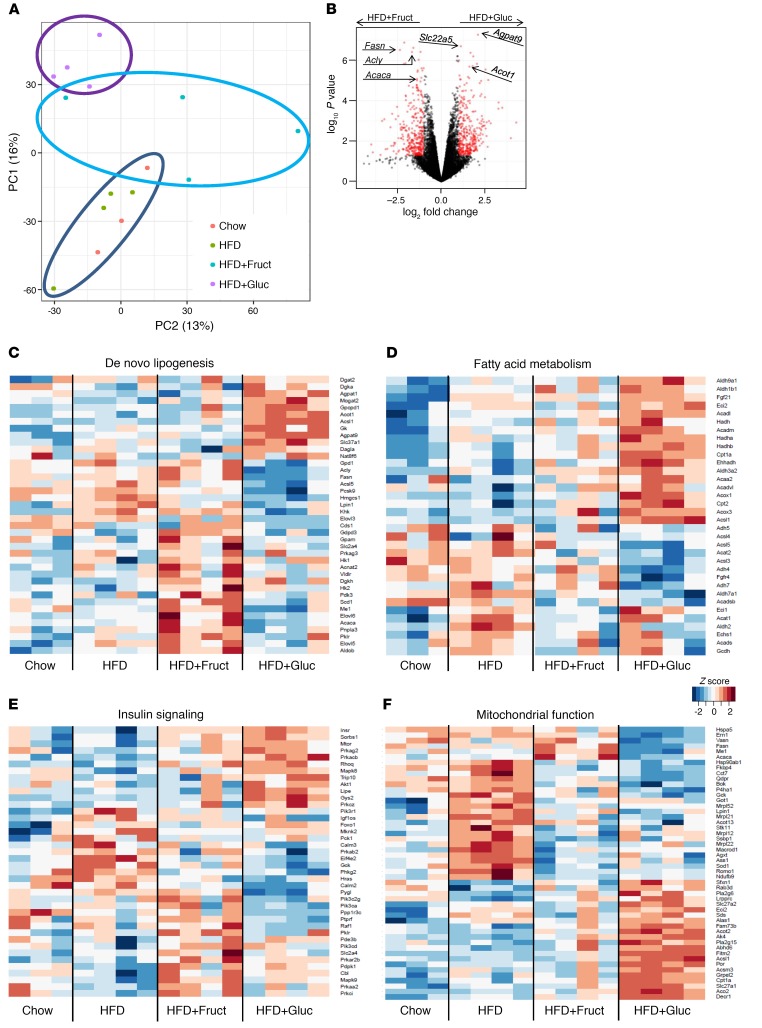
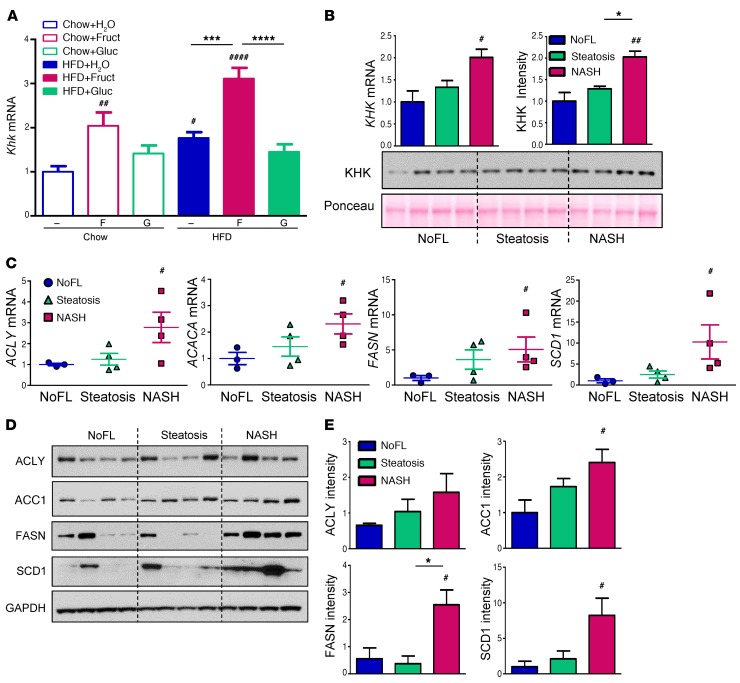
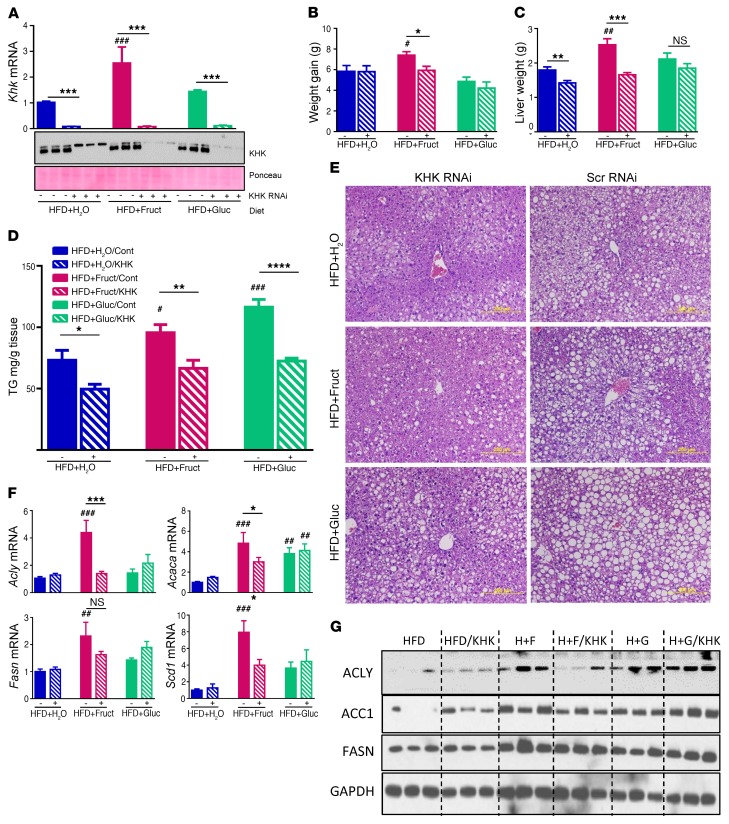
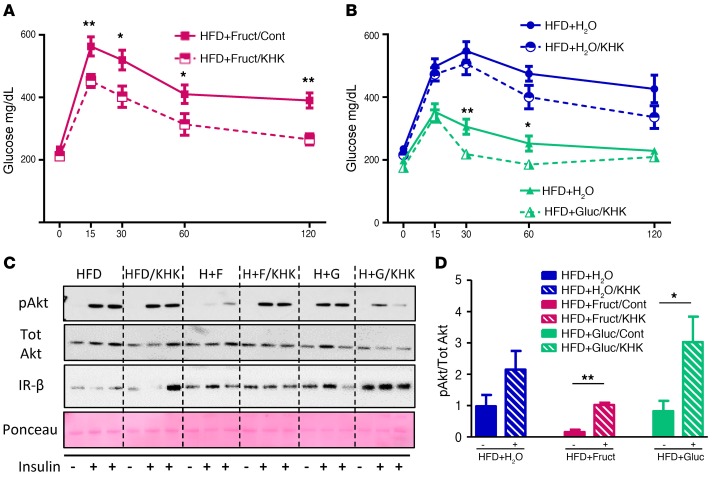
Overconsumption of high-fat diet (HFD) and sugar-sweetened beverages are risk factors for developing obesity, insulin resistance, and fatty liver disease. Here we have dissected mechanisms underlying this association using mice fed either chow or HFD with or without fructose- or glucose-supplemented water. In chow-fed mice, there was no major physiological difference between fructose and glucose supplementation. On the other hand, mice on HFD supplemented with fructose developed more pronounced obesity, glucose intolerance, and hepatomegaly as compared to glucose-supplemented HFD mice, despite similar caloric intake. Fructose and glucose supplementation also had distinct effects on expression of the lipogenic transcription factors ChREBP and SREBP1c. While both sugars increased ChREBP-β, fructose supplementation uniquely increased SREBP1c and downstream fatty acid synthesis genes, resulting in reduced liver insulin signaling. In contrast, glucose enhanced total ChREBP expression and triglyceride synthesis but was associated with improved hepatic insulin signaling. Metabolomic and RNA sequence analysis confirmed dichotomous effects of fructose and glucose supplementation on liver metabolism in spite of inducing similar hepatic lipid accumulation. Ketohexokinase, the first enzyme of fructose metabolism, was increased in fructose-fed mice and in obese humans with steatohepatitis. Knockdown of ketohexokinase in liver improved hepatic steatosis and glucose tolerance in fructose-supplemented mice. Thus, fructose is a component of dietary sugar that is distinctively associated with poor metabolic outcomes, whereas increased glucose intake may be protective.
Conflict of interest statement
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
