

TLR9 stimulation of B-cells induces transcription of p53 and prevents spontaneous and irradiation-induced cell death independent of DNA damage responses. Implications for Common variable immunodeficiency
- PMID: 28973009
- PMCID: PMC5626471
- DOI: 10.1371/journal.pone.0185708
TLR9 stimulation of B-cells induces transcription of p53 and prevents spontaneous and irradiation-induced cell death independent of DNA damage responses. Implications for Common variable immunodeficiency
Abstract
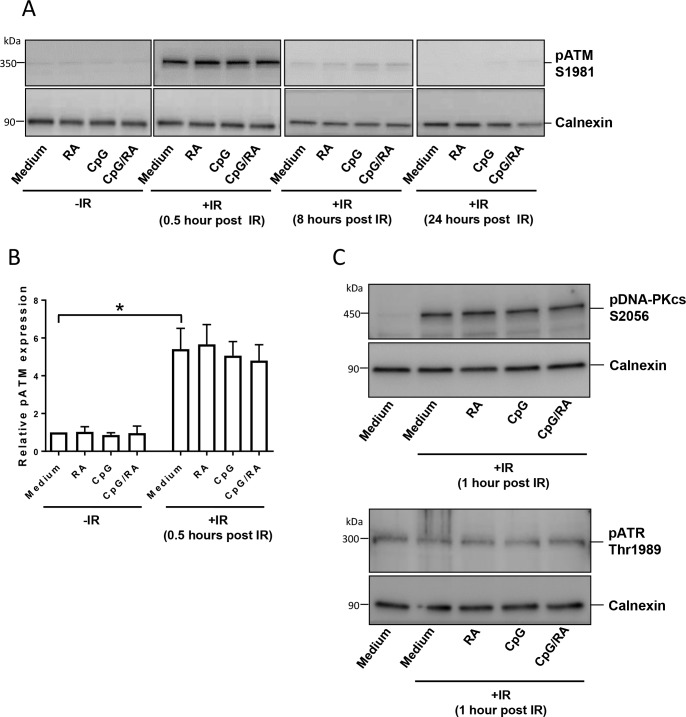
In the present study, we address the important issue of whether B-cells protected from irradiation-induced cell death, may survive with elevated levels of DNA damage. If so, such cells would be at higher risk of gaining mutations and undergoing malignant transformation. We show that stimulation of B-cells with the TLR9 ligands CpG-oligodeoxynucleotides (CpG-ODN) prevents spontaneous and irradiation-induced death of normal peripheral blood B-cells, and of B-cells from patients diagnosed with Common variable immunodeficiency (CVID). The TLR9-mediated survival is enhanced by the vitamin A metabolite retinoic acid (RA). Importantly, neither stimulation of B-cells via TLR9 alone or with RA increases irradiation-induced DNA strand breaks and DNA damage responses such as activation of ATM and DNA-PKcs. We prove that elevated levels of γH2AX imposed by irradiation of stimulated B-cells is not due to induction of DNA double strand breaks, but merely reflects increased levels of total H2AX upon stimulation. Interestingly however, we unexpectedly find that TLR9 stimulation of B-cells induces low amounts of inactive p53, explained by transcriptional induction of TP53. Taken together, we show that enhanced survival of irradiated B-cells is not accompanied by elevated levels of DNA damage. Our results imply that TLR9-mediated activation of B-cells not only promotes cell survival, but may via p53 provide cells with a barrier against harmful consequences of enhanced activation and proliferation. As CVID-derived B-cells are more radiosensitive and prone to undergo apoptosis than normal B-cells, our data support treatment of CVID patients with CpG-ODN and RA.
Conflict of interest statement
Figures




Similar articles
-
Myeloid cell leukaemia 1 has a vital role in retinoic acid-mediated protection of Toll-like receptor 9-stimulated B cells from spontaneous and DNA damage-induced apoptosis.Immunology. 2016 Sep;149(1):62-73. doi: 10.1111/imm.12629. Epub 2016 Jul 25. Immunology. 2016. PMID: 27278254 Free PMC article.
-
Retinoic acid improves defective TLR9/RP105-induced immune responses in common variable immunodeficiency-derived B cells.J Immunol. 2013 Oct 1;191(7):3624-33. doi: 10.4049/jimmunol.1300213. Epub 2013 Sep 4. J Immunol. 2013. PMID: 24006462
-
TLR9 activation is defective in common variable immune deficiency.J Immunol. 2006 Feb 1;176(3):1978-87. doi: 10.4049/jimmunol.176.3.1978. J Immunol. 2006. PMID: 16424230
-
Bovine toll-like receptor 9: a comparative analysis of molecular structure, function and expression.Vet Immunol Immunopathol. 2005 Oct 18;108(1-2):11-6. doi: 10.1016/j.vetimm.2005.07.012. Vet Immunol Immunopathol. 2005. PMID: 16098606 Review.
-
TLR9 as a key receptor for the recognition of DNA.Adv Drug Deliv Rev. 2008 Apr 29;60(7):795-804. doi: 10.1016/j.addr.2007.12.004. Epub 2008 Jan 3. Adv Drug Deliv Rev. 2008. PMID: 18262306 Review.
Cited by
-
TLR9 Polymorphisms Might Contribute to the Ethnicity Bias for EBV-Infected Nasopharyngeal Carcinoma.iScience. 2020 Mar 27;23(3):100937. doi: 10.1016/j.isci.2020.100937. Epub 2020 Feb 28. iScience. 2020. PMID: 32179470 Free PMC article.
-
Heat Killed Salmonella typhimurium Protects Intestine Against Radiation Injury Through Wnt Signaling Pathway.J Oncol. 2021 Jun 18;2021:5550956. doi: 10.1155/2021/5550956. eCollection 2021. J Oncol. 2021. PMID: 34239563 Free PMC article.
-
Multi-omics analysis of naïve B cells of patients harboring the C104R mutation in TACI.Front Immunol. 2022 Aug 16;13:938240. doi: 10.3389/fimmu.2022.938240. eCollection 2022. Front Immunol. 2022. PMID: 36072607 Free PMC article.
-
Heat-killed Salmonella Typhimurium protects mice against carbon ion radiation.J Int Med Res. 2020 Oct;48(10):300060520924256. doi: 10.1177/0300060520924256. J Int Med Res. 2020. PMID: 33021413 Free PMC article.
References
-
- Goldstein M, Kastan MB. The DNA damage response: implications for tumor responses to radiation and chemotherapy. Annu Rev Med 2015;66:129–43. doi: 10.1146/annurev-med-081313-121208 - DOI - PubMed
-
- Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell 2010. October 22;40(2):179–204. doi: 10.1016/j.molcel.2010.09.019 - DOI - PMC - PubMed
-
- Lindahl T. Instability and decay of the primary structure of DNA. Nature 1993. April 22;362(6422):709–15. doi: 10.1038/362709a0 - DOI - PubMed
-
- Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature 2009. October 22;461(7267):1071–8. doi: 10.1038/nature08467 - DOI - PMC - PubMed
-
- Kamran SC, Berrington de GA, Ng A, Haas-Kogan D, Viswanathan AN. Therapeutic radiation and the potential risk of second malignancies. Cancer 2016. June 15;122(12):1809–21. doi: 10.1002/cncr.29841 - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
