

Amyotrophic lateral sclerosis-associated mutant SOD1 inhibits anterograde axonal transport of mitochondria by reducing Miro1 levels
- PMID: 28973175
- PMCID: PMC5886184
- DOI: 10.1093/hmg/ddx348
Amyotrophic lateral sclerosis-associated mutant SOD1 inhibits anterograde axonal transport of mitochondria by reducing Miro1 levels
Abstract
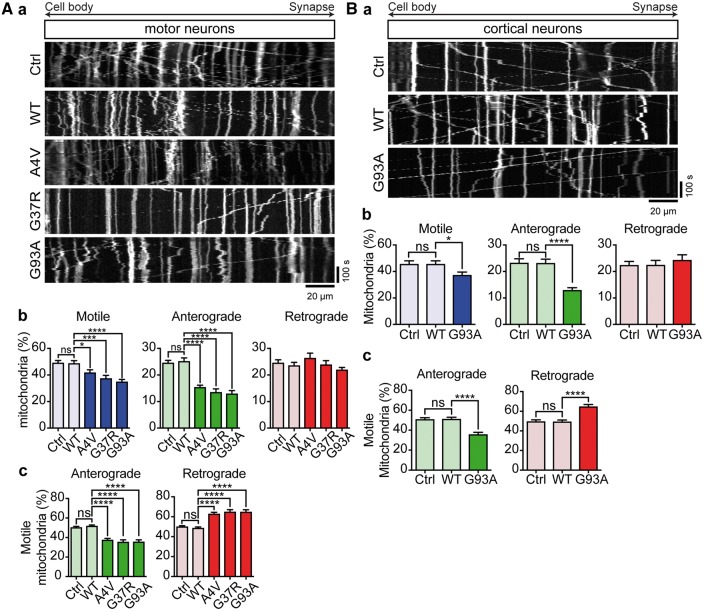
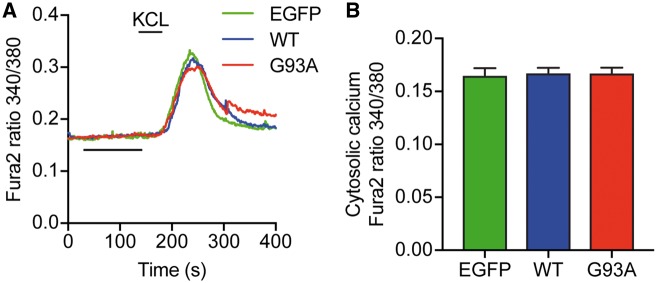
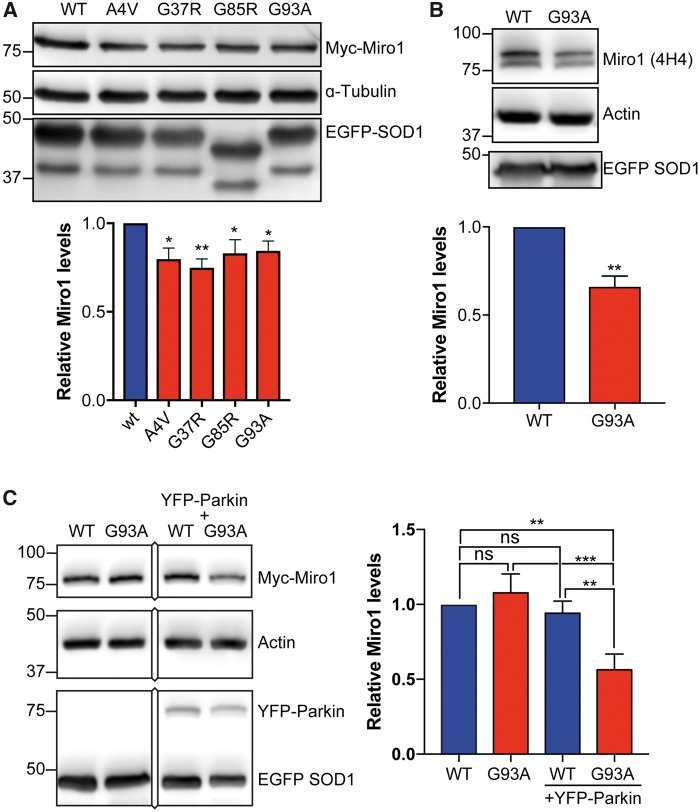
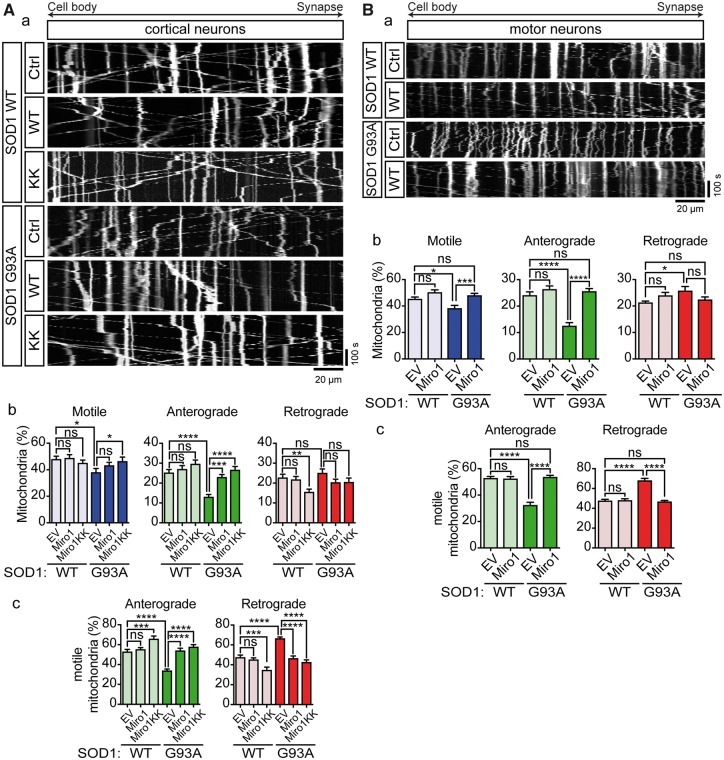
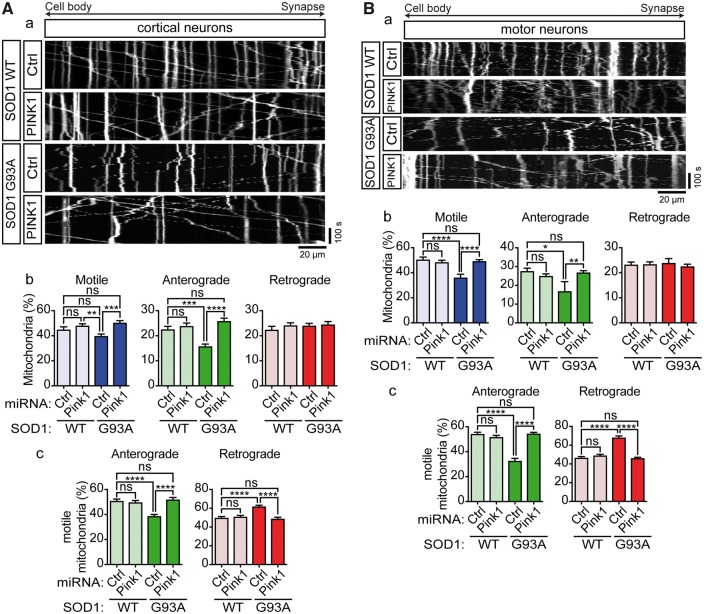
Defective axonal transport is an early neuropathological feature of amyotrophic lateral sclerosis (ALS). We have previously shown that ALS-associated mutations in Cu/Zn superoxide dismutase 1 (SOD1) impair axonal transport of mitochondria in motor neurons isolated from SOD1 G93A transgenic mice and in ALS mutant SOD1 transfected cortical neurons, but the underlying mechanisms remained unresolved. The outer mitochondrial membrane protein mitochondrial Rho GTPase 1 (Miro1) is a master regulator of mitochondrial axonal transport in response to cytosolic calcium (Ca2+) levels ([Ca2+]c) and mitochondrial damage. Ca2+ binding to Miro1 halts mitochondrial transport by modifying its interaction with kinesin-1 whereas mitochondrial damage induces Phosphatase and Tensin Homolog (PTEN)-induced Putative Kinase 1 (PINK1) and Parkin-dependent degradation of Miro1 and consequently stops transport. To identify the mechanism underlying impaired axonal transport of mitochondria in mutant SOD1-related ALS we investigated [Ca2+]c and Miro1 levels in ALS mutant SOD1 expressing neurons. We found that expression of ALS mutant SOD1 reduced the level of endogenous Miro1 but did not affect [Ca2+]c. ALS mutant SOD1 induced reductions in Miro1 levels were Parkin dependent. Moreover, both overexpression of Miro1 and ablation of PINK1 rescued the mitochondrial axonal transport deficit in ALS mutant SOD1-expressing cortical and motor neurons. Together these results provide evidence that ALS mutant SOD1 inhibits axonal transport of mitochondria by inducing PINK1/Parkin-dependent Miro1 degradation.
© The Author 2017. Published by Oxford University Press.
Figures
References
-
- Kiernan M.C., Vucic S., Cheah B.C., Turner M.R., Eisen A., Hardiman O., Burrell J.R., Zoing M.C. (2011) Amyotrophic lateral sclerosis. Lancet, 377, 942–955. - PubMed
-
- Abel O., Powell J.F., Andersen P.M., Al-Chalabi A. (2012) ALSoD: a user-friendly online bioinformatics tool for amyotrophic lateral sclerosis genetics. Hum. Mutat., 33, 1345–1351. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
