

TUBB4A mutations result in specific neuronal and oligodendrocytic defects that closely match clinically distinct phenotypes
- PMID: 28973395
- PMCID: PMC7462055
- DOI: 10.1093/hmg/ddx338
TUBB4A mutations result in specific neuronal and oligodendrocytic defects that closely match clinically distinct phenotypes
Abstract
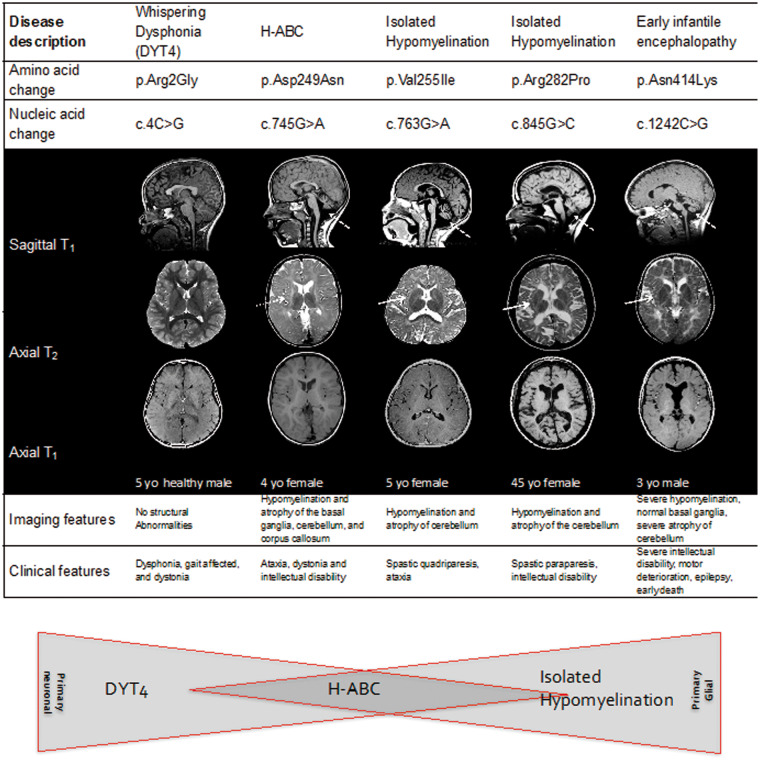
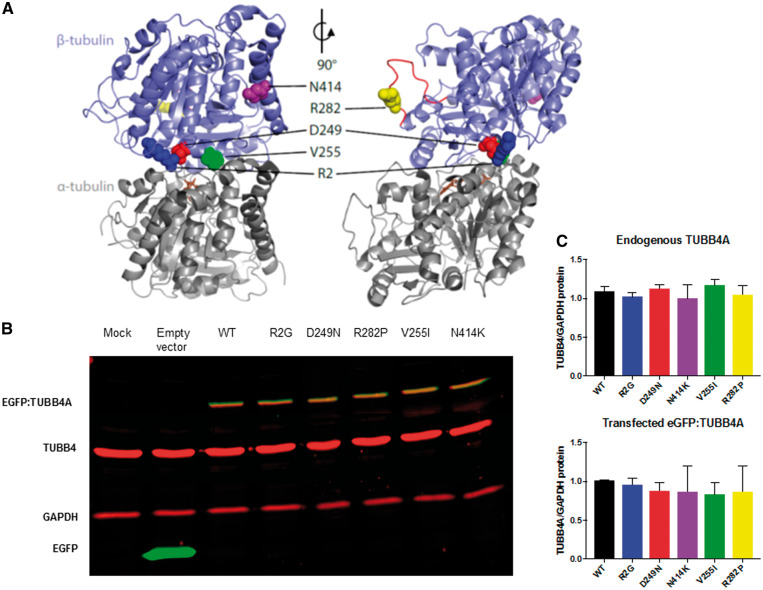
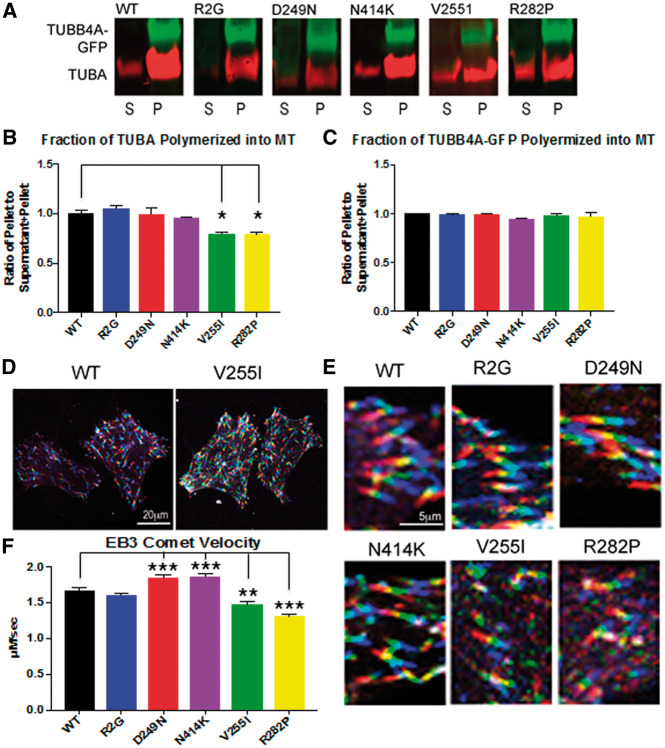
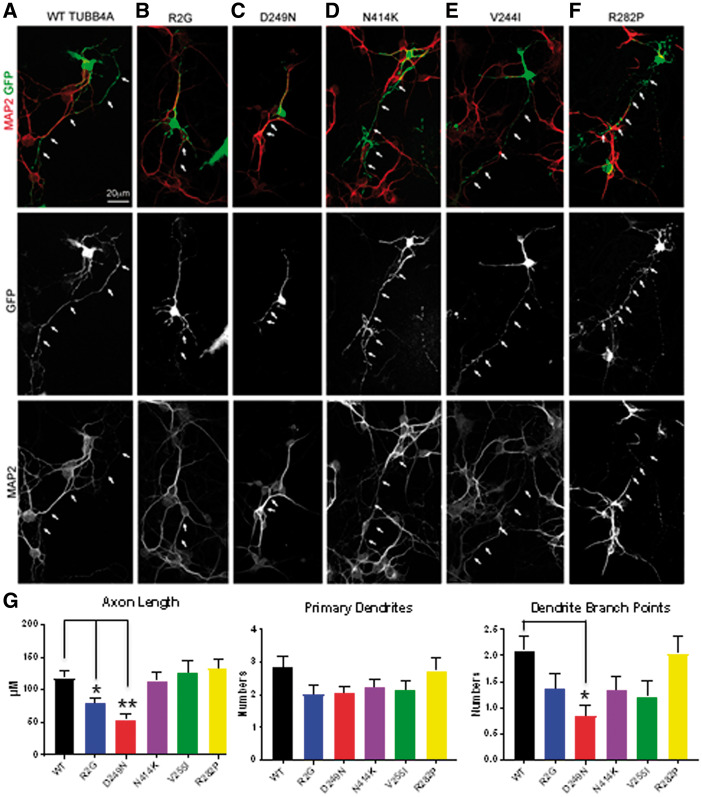
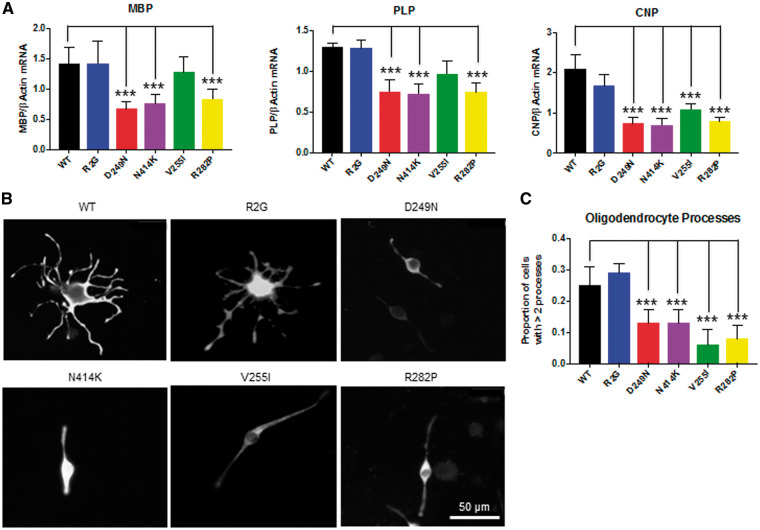
Hypomyelinating leukodystrophies are heritable disorders defined by lack of development of brain myelin, but the cellular mechanisms of hypomyelination are often poorly understood. Mutations in TUBB4A, encoding the tubulin isoform tubulin beta class IVA (Tubb4a), result in the symptom complex of hypomyelination with atrophy of basal ganglia and cerebellum (H-ABC). Additionally, TUBB4A mutations are known to result in a broad phenotypic spectrum, ranging from primary dystonia (DYT4), isolated hypomyelination with spastic quadriplegia, and an infantile onset encephalopathy, suggesting multiple cell types may be involved. We present a study of the cellular effects of TUBB4A mutations responsible for H-ABC (p.Asp249Asn), DYT4 (p.Arg2Gly), a severe combined phenotype with hypomyelination and encephalopathy (p.Asn414Lys), as well as milder phenotypes causing isolated hypomyelination (p.Val255Ile and p.Arg282Pro). We used a combination of histopathological, biochemical and cellular approaches to determine how these different mutations may have variable cellular effects in neurons and/or oligodendrocytes. Our results demonstrate that specific mutations lead to either purely neuronal, combined neuronal and oligodendrocytic or purely oligodendrocytic defects that closely match their respective clinical phenotypes. Thus, the DYT4 mutation that leads to phenotypes attributable to neuronal dysfunction results in altered neuronal morphology, but with unchanged tubulin quantity and polymerization, with normal oligodendrocyte morphology and myelin gene expression. Conversely, mutations associated with isolated hypomyelination (p.Val255Ile and p.Arg282Pro) and the severe combined phenotype (p.Asn414Lys) resulted in normal neuronal morphology but were associated with altered oligodendrocyte morphology, myelin gene expression, and microtubule dysfunction. The H-ABC mutation (p.Asp249Asn) that exhibits a combined neuronal and myelin phenotype had overlapping cellular defects involving both neuronal and oligodendrocyte cell types in vitro. Only mutations causing hypomyelination phenotypes showed altered microtubule dynamics and acted through a dominant toxic gain of function mechanism. The DYT4 mutation had no impact on microtubule dynamics suggesting a distinct mechanism of action. In summary, the different clinical phenotypes associated with TUBB4A reflect the selective and specific cellular effects of the causative mutations. Cellular specificity of disease pathogenesis is relevant to developing targeted treatments for this disabling condition.
© The Author 2017. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures

References
-
- Hamilton E.M., Polder E., Vanderver A., Naidu S., Schiffmann R., Fisher K., Raguž A.B., Blumkin L., van Berkel C.G.M., Waisfisz Q.. et al. (2014) Hypomyelination with atrophy of the basal ganglia and cerebellum: further delineation of the phenotype and genotype-phenotype correlation. Brain, 137, 1921–1930. - PMC - PubMed
-
- Kumar K.R., Vulinovic F., Lohmann K., Park J.S., Schaake S., Sue C.M., Klein C. (2015) Mutations in TUBB4A and spastic paraplegia. Mov. Disord., 30, 1857–1858. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
