

Modulation of proteostasis counteracts oxidative stress and affects DNA base excision repair capacity in ATM-deficient cells
- PMID: 28973444
- PMCID: PMC5622344
- DOI: 10.1093/nar/gkx635
Modulation of proteostasis counteracts oxidative stress and affects DNA base excision repair capacity in ATM-deficient cells
Abstract
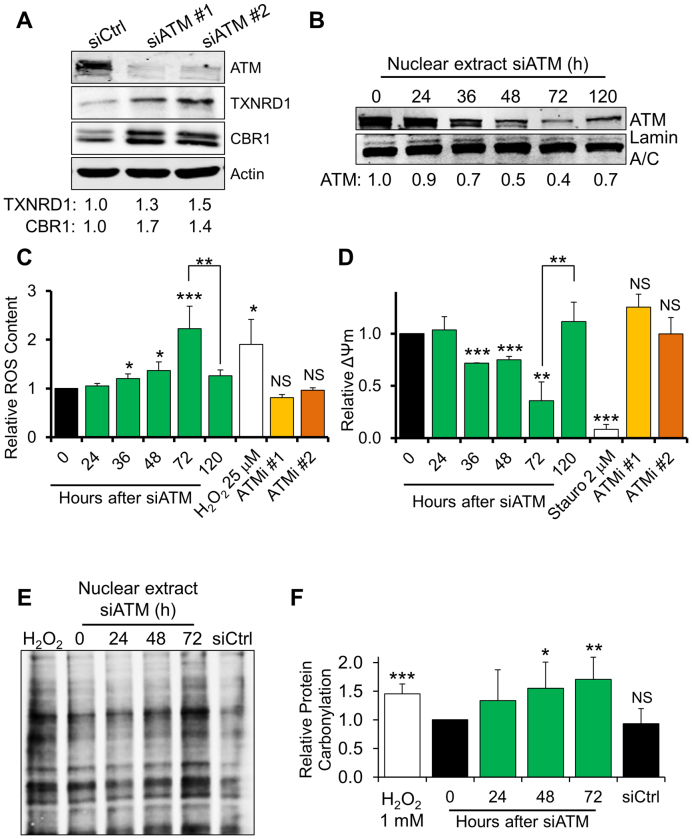
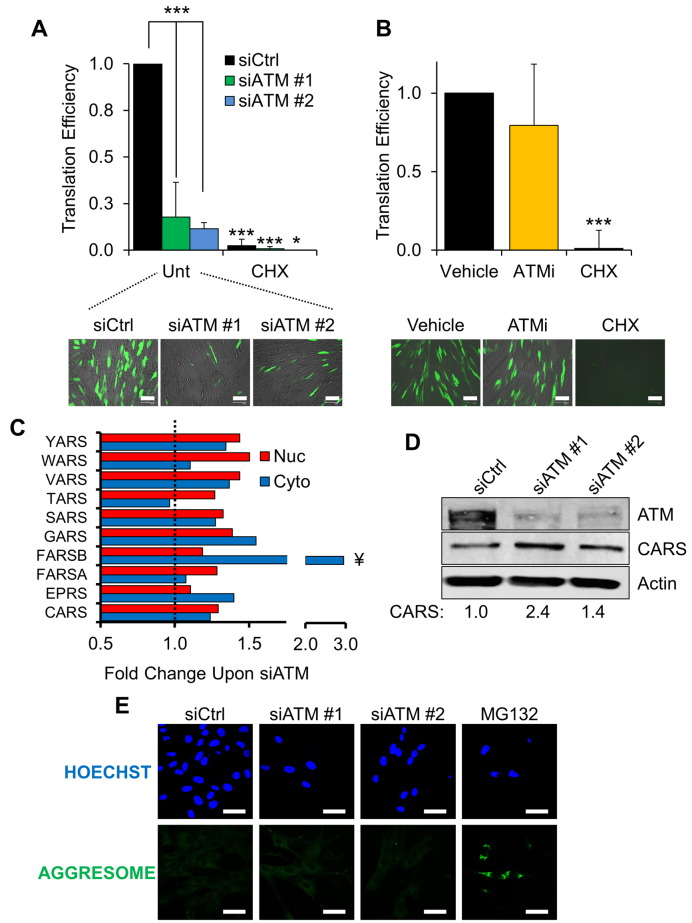
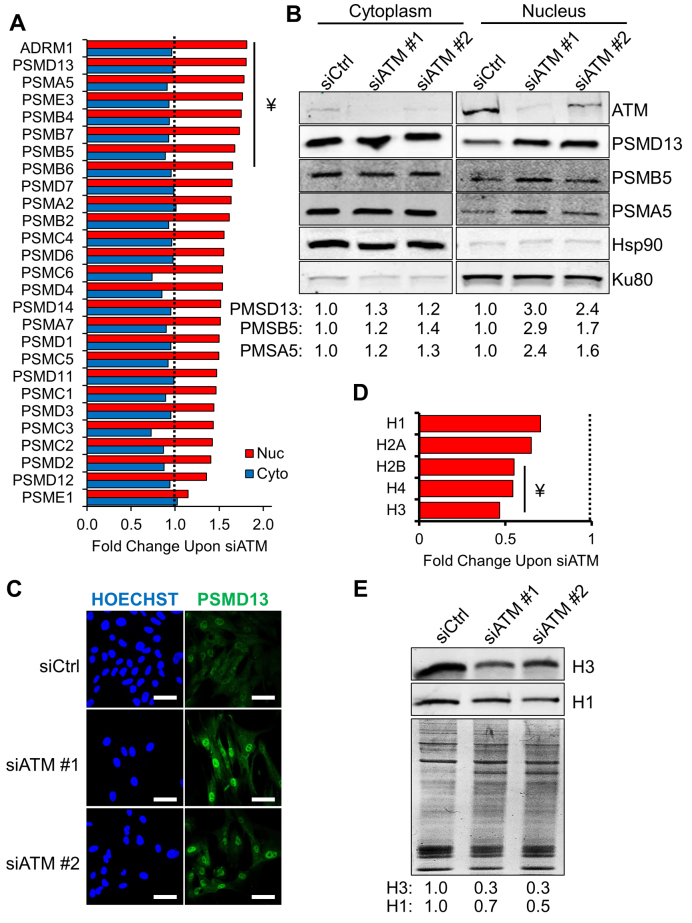
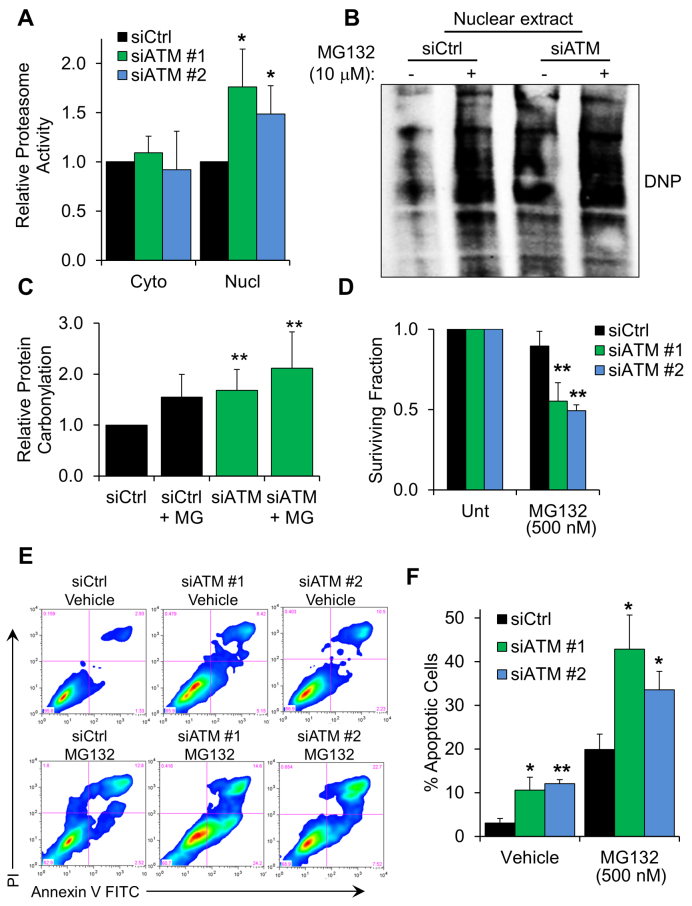
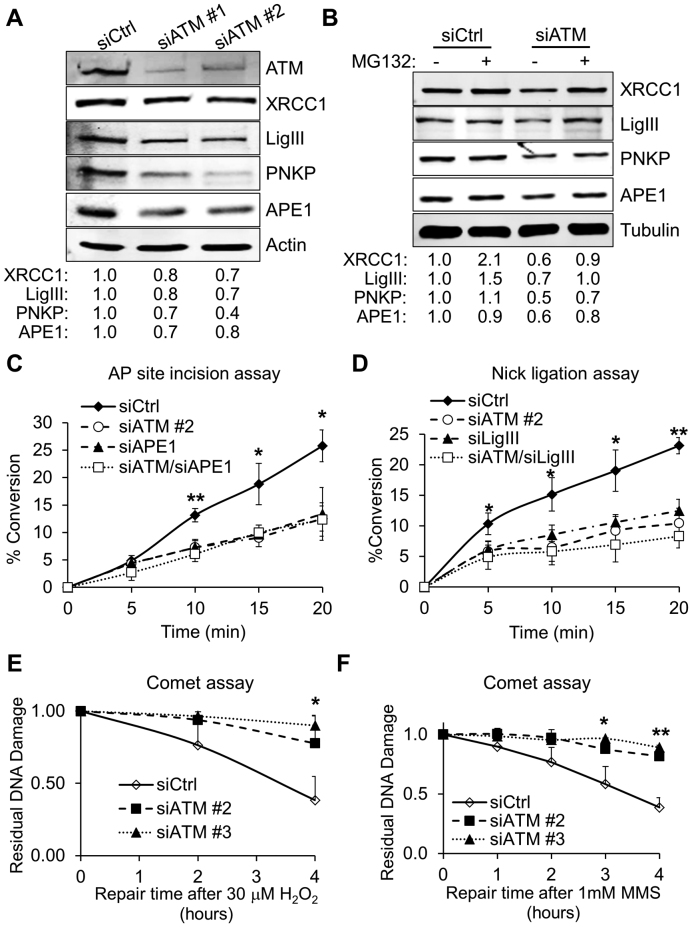
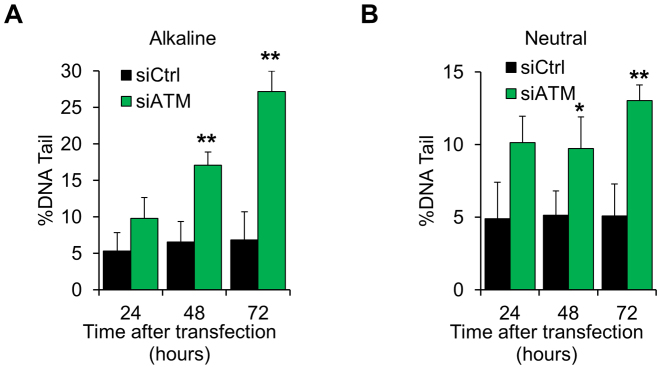
Ataxia telangiectasia (A-T) is a syndrome associated with loss of ATM protein function. Neurodegeneration and cancer predisposition, both hallmarks of A-T, are likely to emerge as a consequence of the persistent oxidative stress and DNA damage observed in this disease. Surprisingly however, despite these severe features, a lack of functional ATM is still compatible with early life, suggesting that adaptation mechanisms contributing to cell survival must be in place. Here we address this gap in our knowledge by analysing the process of human fibroblast adaptation to the lack of ATM. We identify profound rearrangement in cellular proteostasis occurring very early on after loss of ATM in order to counter protein damage originating from oxidative stress. Change in proteostasis, however, is not without repercussions. Modulating protein turnover in ATM-depleted cells also has an adverse effect on the DNA base excision repair pathway, the major DNA repair system that deals with oxidative DNA damage. As a consequence, the burden of unrepaired endogenous DNA lesions intensifies, progressively leading to genomic instability. Our study provides a glimpse at the cellular consequences of loss of ATM and highlights a previously overlooked role for proteostasis in maintaining cell survival in the absence of ATM function.
© The Author(s) 2017. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
Similar articles
-
Cellular functions of the protein kinase ATM and their relevance to human disease.Nat Rev Mol Cell Biol. 2021 Dec;22(12):796-814. doi: 10.1038/s41580-021-00394-2. Epub 2021 Aug 24. Nat Rev Mol Cell Biol. 2021. PMID: 34429537 Review.
-
Modulation of hypersensitivity to oxidative DNA damage in ATM defective cells induced by potassium bromate by inhibition of the Poly (ADP-ribose) polymerase (PARP).Mutat Res Genet Toxicol Environ Mutagen. 2018 Dec;836(Pt A):117-123. doi: 10.1016/j.mrgentox.2018.05.009. Epub 2018 May 8. Mutat Res Genet Toxicol Environ Mutagen. 2018. PMID: 30389154
-
Oxidative stress and the multifaceted roles of ATM in maintaining cellular redox homeostasis.Redox Biol. 2024 Sep;75:103269. doi: 10.1016/j.redox.2024.103269. Epub 2024 Jul 16. Redox Biol. 2024. PMID: 39018798 Free PMC article. Review.
-
Role of DDX1 in the oxidative response of ataxia telangiectasia patient-derived fibroblasts.Redox Biol. 2024 Feb;69:102988. doi: 10.1016/j.redox.2023.102988. Epub 2023 Dec 9. Redox Biol. 2024. PMID: 38096740 Free PMC article.
-
Ataxia-telangiectasia-mutated-dependent activation of Ku in human fibroblasts exposed to hydrogen peroxide.Ann N Y Acad Sci. 2006 Dec;1091:76-82. doi: 10.1196/annals.1378.056. Ann N Y Acad Sci. 2006. PMID: 17341604
Cited by
-
Quercetin enhances stress resistance in Saccharomyces cerevisiae tel1 mutant cells to different stressors.J Food Sci Technol. 2018 Apr;55(4):1455-1466. doi: 10.1007/s13197-018-3062-9. Epub 2018 Feb 19. J Food Sci Technol. 2018. PMID: 29606760 Free PMC article.
-
SIRT7 Downregulation Promotes Breast Cancer Metastasis Via LAP2α-Induced Chromosomal Instability.Int J Biol Sci. 2023 Mar 5;19(5):1528-1542. doi: 10.7150/ijbs.75340. eCollection 2023. Int J Biol Sci. 2023. PMID: 37056924 Free PMC article.
-
Metabolic Stress and Mitochondrial Dysfunction in Ataxia-Telangiectasia.Antioxidants (Basel). 2022 Mar 28;11(4):653. doi: 10.3390/antiox11040653. Antioxidants (Basel). 2022. PMID: 35453338 Free PMC article. Review.
-
Cellular functions of the protein kinase ATM and their relevance to human disease.Nat Rev Mol Cell Biol. 2021 Dec;22(12):796-814. doi: 10.1038/s41580-021-00394-2. Epub 2021 Aug 24. Nat Rev Mol Cell Biol. 2021. PMID: 34429537 Review.
-
When nature's robots go rogue: exploring protein homeostasis dysfunction and the implications for understanding human aging disease pathologies.Expert Rev Proteomics. 2018 Apr;15(4):293-309. doi: 10.1080/14789450.2018.1453362. Epub 2018 Mar 21. Expert Rev Proteomics. 2018. PMID: 29540077 Free PMC article. Review.
References
-
- Lavin M.F., Gueven N., Bottle S., Gatti R.A.. Current and potential therapeutic strategies for the treatment of ataxia-telangiectasia. Br. Med. Bull. 2007; 81–82:129–147. - PubMed
-
- Savitsky K., Bar-Shira A., Gilad S., Rotman G., Ziv Y., Vanagaite L., Tagle D.A., Smith S., Uziel T., Sfez S. et al. . A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995; 268:1749–1753. - PubMed
-
- Gatti R.A., Berkel I., Boder E., Braedt G., Charmley P., Concannon P., Ersoy F., Foroud T., Jaspers N.G., Lange K. et al. . Localization of an ataxia-telangiectasia gene to chromosome 11q22-23. Nature. 1988; 336:577–580. - PubMed
-
- Paull T.T. Mechanisms of ATM Activation. Annu. Rev. Biochem. 2015; 84:711–738. - PubMed
-
- Bakkenist C.J., Kastan M.B.. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003; 421:499–506. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
