

A human patient-derived cellular model of Joubert syndrome reveals ciliary defects which can be rescued with targeted therapies
- PMID: 28973549
- PMCID: PMC5886250
- DOI: 10.1093/hmg/ddx347
A human patient-derived cellular model of Joubert syndrome reveals ciliary defects which can be rescued with targeted therapies
Abstract
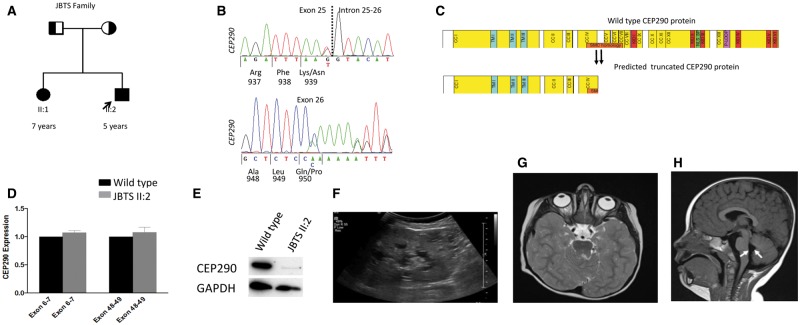
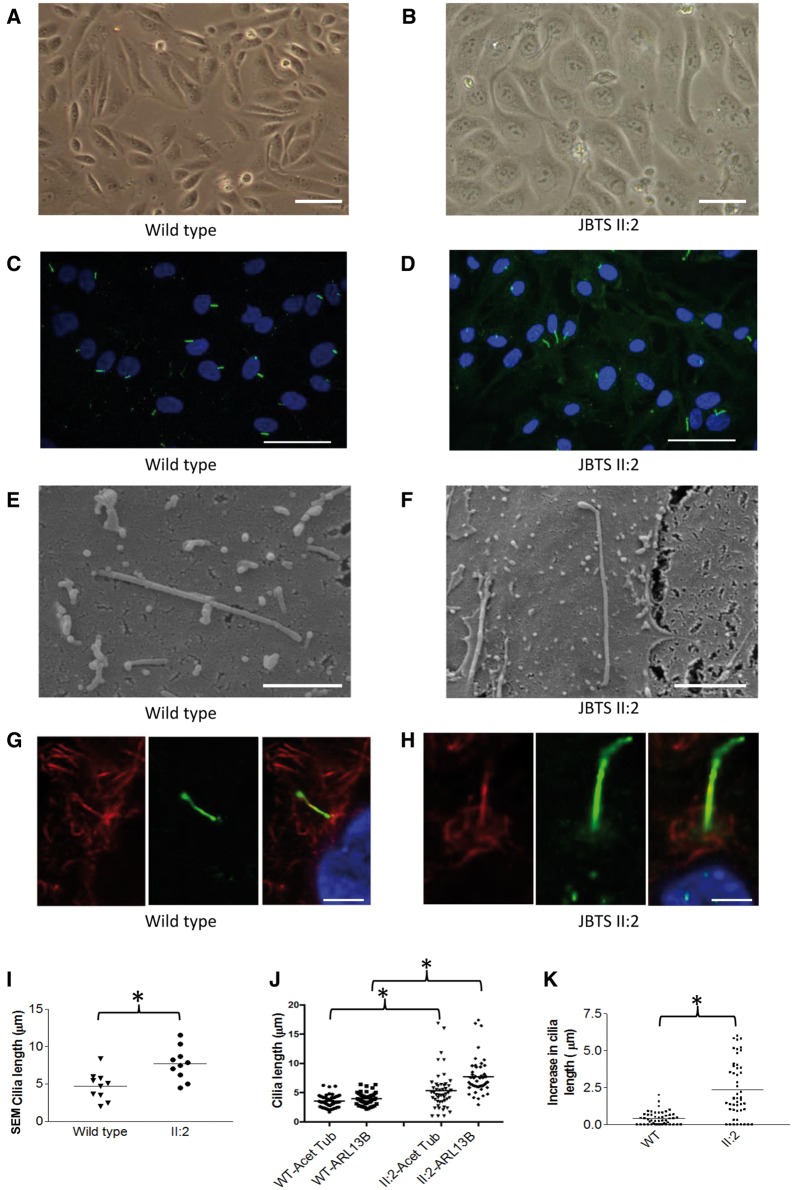
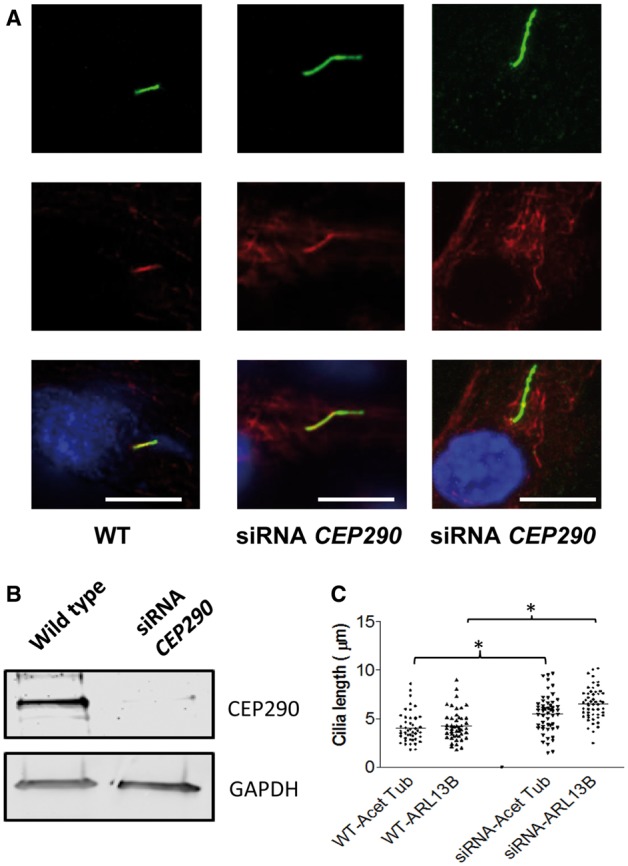
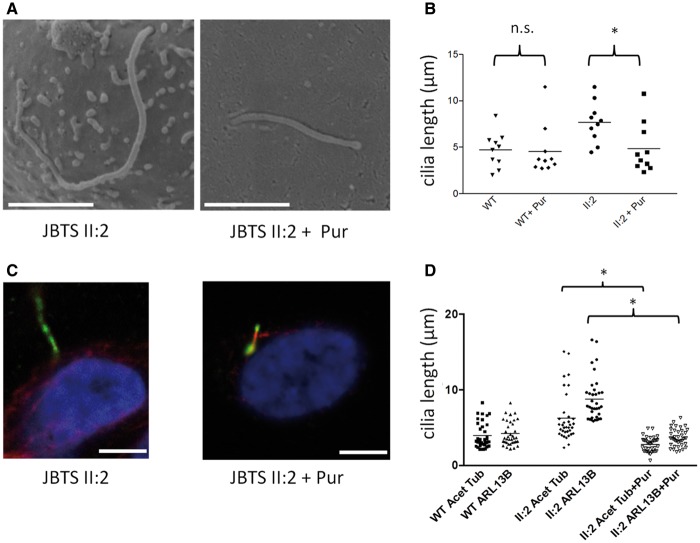
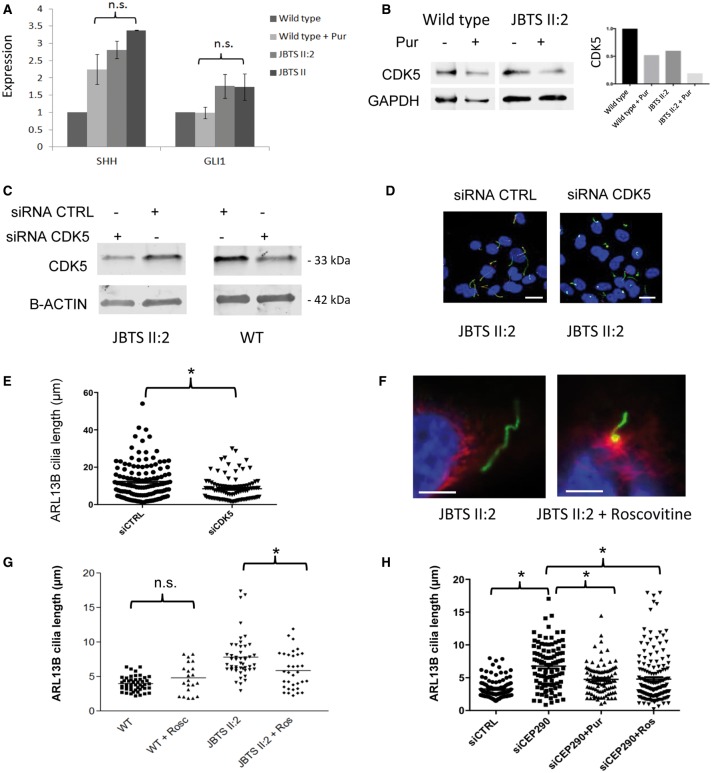
Joubert syndrome (JBTS) is the archetypal ciliopathy caused by mutation of genes encoding ciliary proteins leading to multi-system phenotypes, including a cerebello-retinal-renal syndrome. JBTS is genetically heterogeneous, however mutations in CEP290 are a common underlying cause. The renal manifestation of JBTS is a juvenile-onset cystic kidney disease, known as nephronophthisis, typically progressing to end-stage renal failure within the first two decades of life, thus providing a potential window for therapeutic intervention. In order to increase understanding of JBTS and its associated kidney disease and to explore potential treatments, we conducted a comprehensive analysis of primary renal epithelial cells directly isolated from patient urine (human urine-derived renal epithelial cells, hURECs). We demonstrate that hURECs from a JBTS patient with renal disease have elongated and disorganized primary cilia and that this ciliary phenotype is specifically associated with an absence of CEP290 protein. Treatment with the Sonic hedgehog (Shh) pathway agonist purmorphamine or cyclin-dependent kinase inhibition (using roscovitine and siRNA directed towards cyclin-dependent kinase 5) ameliorated the cilia phenotype. In addition, purmorphamine treatment was shown to reduce cyclin-dependent kinase 5 in patient cells, suggesting a convergence of these signalling pathways. To our knowledge, this is the most extensive analysis of primary renal epithelial cells from JBTS patients to date. It demonstrates the feasibility and power of this approach to directly assess the consequences of patient-specific mutations in a physiologically relevant context and a previously unrecognized convergence of Shh agonism and cyclin-dependent kinase inhibition as potential therapeutic targets.
© The Author 2017. Published by Oxford University Press.
Figures
References
-
- Sayer J.A., Otto E.A., O'Toole J.F., Nurnberg G., Kennedy M.A., Becker C., Hennies H.C., Helou J., Attanasio M., Fausett B.V.. et al. (2006) The centrosomal protein nephrocystin-6 is mutated in Joubert syndrome and activates transcription factor ATF4. Nat. Genet., 38, 674–681. - PubMed
-
- Valente E.M., Silhavy J.L., Brancati F., Barrano G., Krishnaswami S.R., Castori M., Lancaster M.A., Boltshauser E., Boccone L., Al-Gazali L.. et al. (2006) Mutations in CEP290, which encodes a centrosomal protein, cause pleiotropic forms of Joubert syndrome. Nat. Genet., 38, 623–625. - PubMed
-
- Perrault I., Delphin N., Hanein S., Gerber S., Dufier J.L., Roche O., Defoort-Dhellemmes S., Dollfus H., Fazzi E., Munnich A.. et al. (2007) Spectrum of NPHP6/CEP290 mutations in Leber congenital amaurosis and delineation of the associated phenotype. Hum. Mutat., 28, 416. - PubMed
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
