

Granulocyte-colony-stimulating factor (G-CSF) signaling in spinal microglia drives visceral sensitization following colitis
- PMID: 28973941
- PMCID: PMC5651747
- DOI: 10.1073/pnas.1706053114
Granulocyte-colony-stimulating factor (G-CSF) signaling in spinal microglia drives visceral sensitization following colitis
Abstract
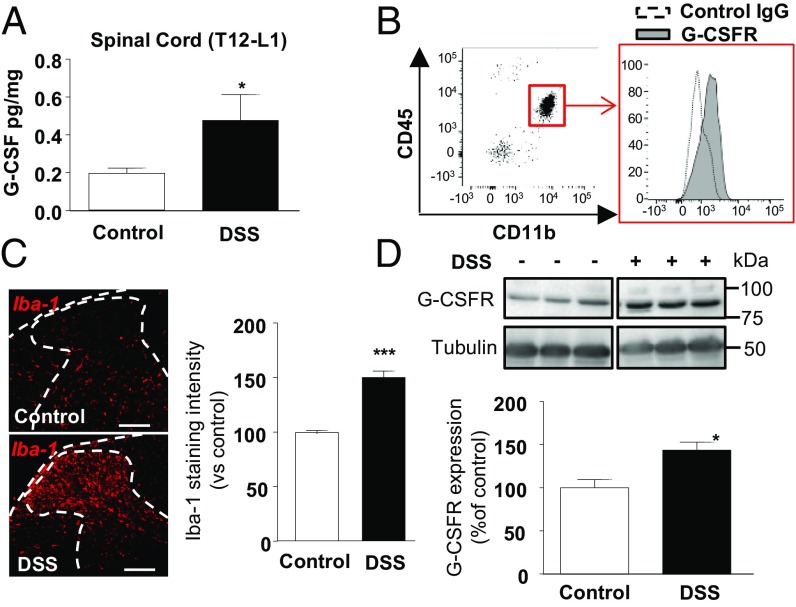
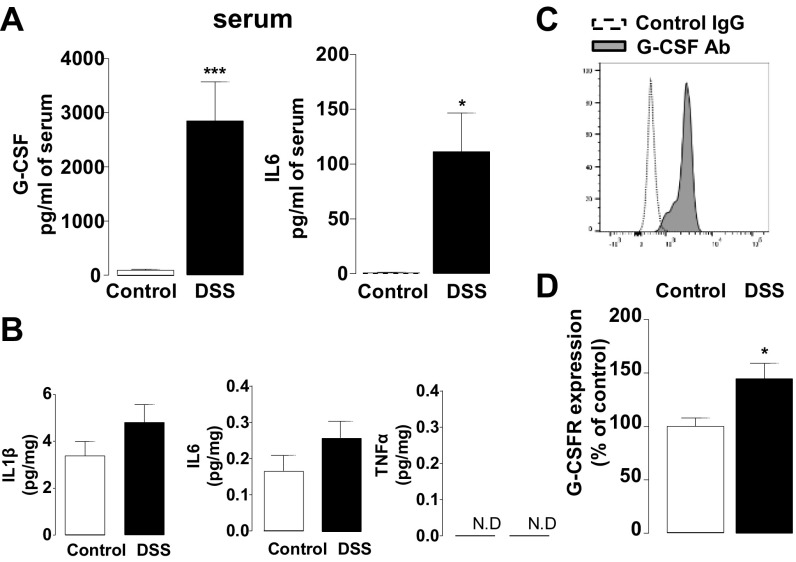
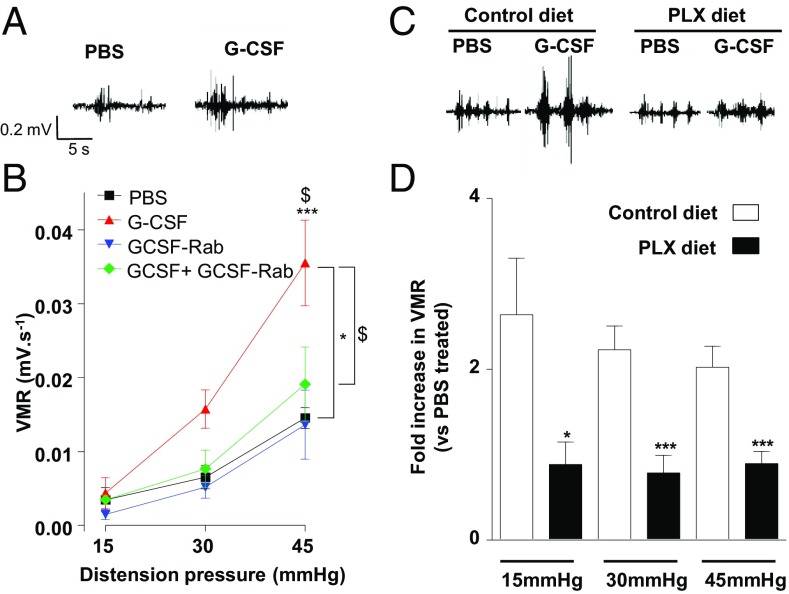
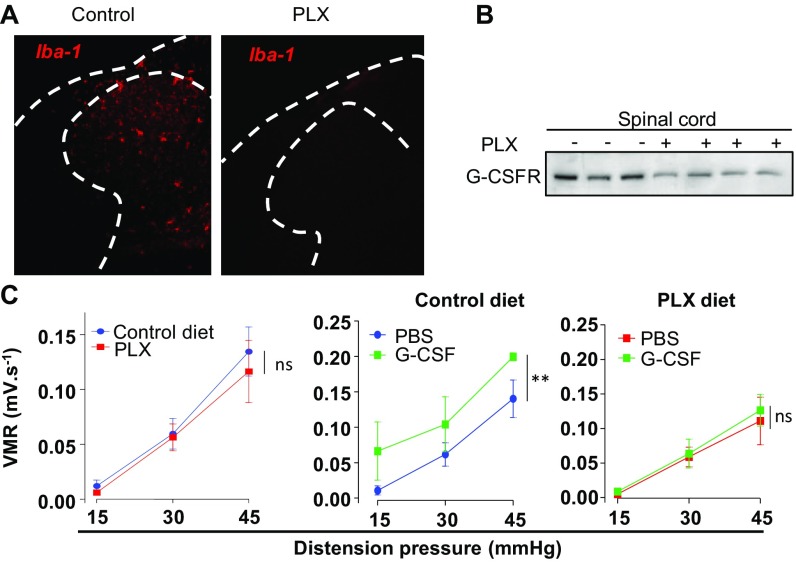
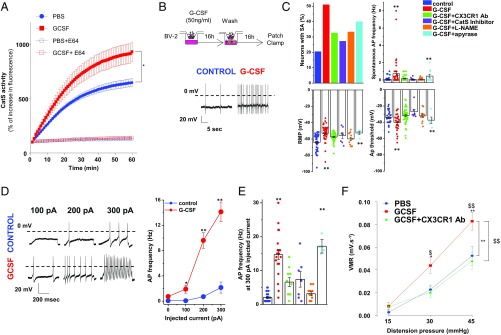
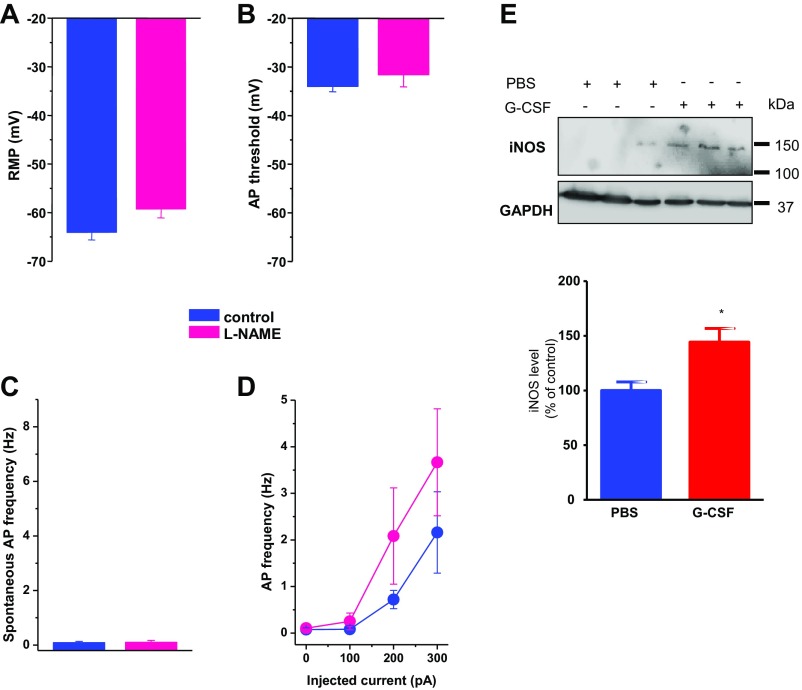
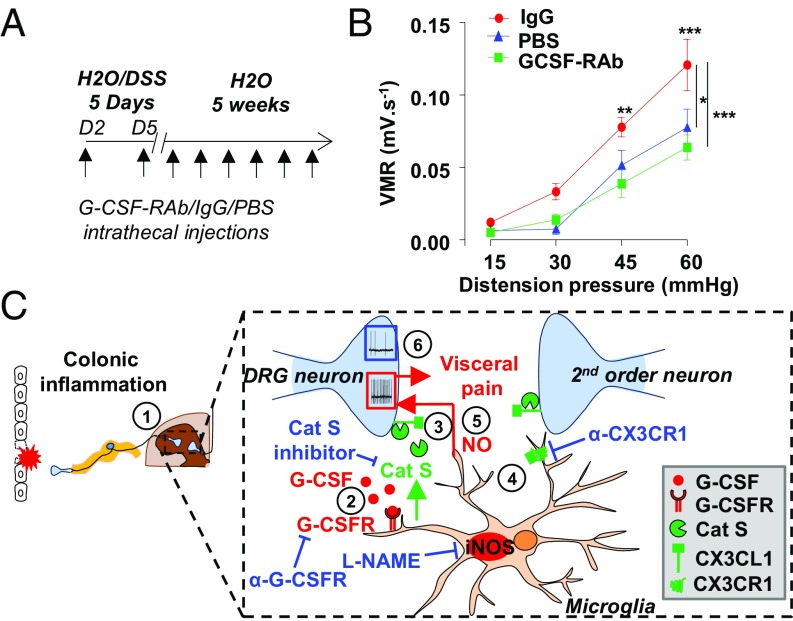
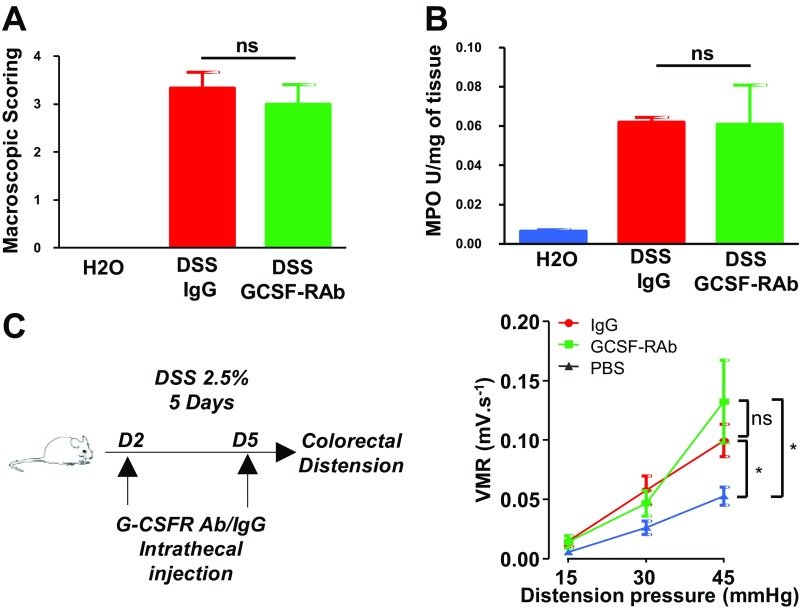
Pain is a main symptom of inflammatory diseases and often persists beyond clinical remission. Although we have a good understanding of the mechanisms of sensitization at the periphery during inflammation, little is known about the mediators that drive central sensitization. Recent reports have identified hematopoietic colony-stimulating factors as important regulators of tumor- and nerve injury-associated pain. Using a mouse model of colitis, we identify the proinflammatory cytokine granulocyte-colony-stimulating factor (G-CSF or Csf-3) as a key mediator of visceral sensitization. We report that G-CSF is specifically up-regulated in the thoracolumbar spinal cord of colitis-affected mice. Our results show that resident spinal microglia express the G-CSF receptor and that G-CSF signaling mediates microglial activation following colitis. Furthermore, healthy mice subjected to intrathecal injection of G-CSF exhibit pronounced visceral hypersensitivity, an effect that is abolished by microglial depletion. Mechanistically, we demonstrate that G-CSF injection increases Cathepsin S activity in spinal cord tissues. When cocultured with microglia BV-2 cells exposed to G-CSF, dorsal root ganglion (DRG) nociceptors become hyperexcitable. Blocking CX3CR1 or nitric oxide production during G-CSF treatment reduces excitability and G-CSF-induced visceral pain in vivo. Finally, administration of G-CSF-neutralizing antibody can prevent the establishment of persistent visceral pain postcolitis. Overall, our work uncovers a DRG neuron-microglia interaction that responds to G-CSF by engaging Cathepsin S-CX3CR1-inducible NOS signaling. This interaction represents a central step in visceral sensitization following colonic inflammation, thereby identifying spinal G-CSF as a target for treating chronic abdominal pain.
Keywords: G-CSF; colitis; microglia; nociceptors; visceral pain.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Basso L, Bourreille A, Dietrich G. Intestinal inflammation and pain management. Curr Opin Pharmacol. 2015;25:50–55. - PubMed
-
- Farrokhyar F, Marshall JK, Easterbrook B, Irvine EJ. Functional gastrointestinal disorders and mood disorders in patients with inactive inflammatory bowel disease: Prevalence and impact on health. Inflamm Bowel Dis. 2006;12:38–46. - PubMed
-
- Young Casey C, Greenberg MA, Nicassio PM, Harpin RE, Hubbard D. Transition from acute to chronic pain and disability: A model including cognitive, affective, and trauma factors. Pain. 2008;134:69–79. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
